AUCTORES
Globalize your Research

Research Article | DOI: https://doi.org/10.31579/2690-1897/235
1Department of Chemistry and Toxins, Judicial Expertise and Research Center, Tripoli, Libya.
2Department of Medicinal Chemistry, Faculty of Pharmacy, University of Tripoli, Tripoli, Libya.
3Department of Biosciences, University of Salzburg, Salzburg, Austria.
*Corresponding Author: Abdul M Gbaj, Professor of Genetics and Biochemistry, Department of Medicinal Chemistry,Faculty of Pharmacy, University of Tripoli, Tripoli, Libya.
Citation: Sofian S. Mohamed, Salah M Bensaber, Nisreen H Meiqal, Anton Hermann, Abdul M Gbaj. (2025). Design And In-Silico Evaluation of Pyridine-4-Carbohydrazide Derivatives for Potential Therapeutic Applications, J, Surgical Case Reports and Images, 8(3); DOI:10.31579/2690-1897/235
Copyright: © 2025, Abdul M Gbaj. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 10 February 2025 | Accepted: 26 February 2025 | Published: 05 March 2025
Keywords: drug discovery; drug repositioning; in-silico study; adme properties
The emergence of drug-resistant and novel diseases underscores the urgency for innovative therapeutic interventions. Drug repositioning and computational approaches offer an efficient pathway to accelerate drug discovery and development. This study leverages these techniques in designing and evaluating derivatives based on the FDA-approved compound, pyridine-4-carbohydrazide, to assess how structural modifications impact therapeutic potential.
Methods: The derivatives were designed using a chemical library of small molecules containing imine functional groups, built upon pyridine-4-carbohydrazide scaffolds (INH01-INH19). Computational tools, including Molinspiration Cheminformatics, way2drug, and the pkCSM platform, were utilized to evaluate each derivative's physicochemical properties, drug-likeness, bioactivity scores, potential biological activities, and ADME (Absorption, Distribution, Metabolism, Excretion) profiles.
Results: Most derivatives demonstrated enhanced physicochemical characteristics, adhering to both Lipinski’s Rule of Five and Veber’s Rule. Bioactivity scores varied with moderate to inactive interactions across six target classes, ranked as follows: enzyme inhibitors, kinase inhibitors, G-protein-coupled receptors, protease inhibitors, nuclear receptors, and ion channel modulators. Notably, derivatives INH03, INH09, INH14, and INH19 exhibited high predicted activity in multiple therapeutic areas, indicating potential applications in antibacterial, antiviral, antiprotozoal, anti-inflammatory and anticancer treatments. Moreover, structural modifications in these derivatives positively influenced ADME profiles compared to pyridine-4-carbohydrazide, though certain compounds presented challenges, such as limited solubility, P-glycoprotein interactions and CYP450 inhibition.
Conclusions: These Schiff base derivatives stand out as promising candidates for further drug development, underscoring the importance of computational strategies in optimizing drug discovery and design.
A drug, in the context of medicine, is a chemical or biological substance designed to interact with biological processes within an organism to treat, diagnose, or prevent disease (Benedetti, 2014). These agents can originate from diverse sources, including natural, semisynthetic, or synthetic origins(Pollock et al., 2024). The continuous need for new medications arises from several factors, including the toxicity and side effects of existing drugs(Kroschinsky et al., 2017), the emergence of new diseases(Khan et al., 2020), the development of drug resistance(Jackson et al., 2018), and advancements in our understanding of health conditions(Subbiah, 2023). These challenges actuate pharmaceutical research and development to innovate, improve treatment efficacy and address previously unmet medical needs. The traditional process of drug discovery follows a well-established procedure, beginning with the identification and validation of molecular targets(Salazar & Gormley, 2017). This is typically followed by high-throughput screening of extensive chemical libraries to identify potential lead compounds(Sinha &Vohora, 2017). Conventional drug discovery methods heavily rely on empirical approaches, involving extensive in vitro and in vivo testing to evaluate a compound's efficacy, pharmacokinetics and toxicity. Although this approach has led to the development of numerous successful therapies, it is both time-consuming and costly, often requiring over a decade and more than $2.5 billion to successfully bring a new drug to market(Kumari et al., 2022; Schlander et al., 2021). To overcome these challenges, alternative approaches such as drug repurposing, also known as drug repositioning, have gained prominence(Ahmad et al., 2021; Talevi& Bellera, 2020a). Drug repositioning involves identifying new therapeutic uses for existing drugs, whether FDA-approved, withdrawn, or outdated. This approach leverages the well-established safety profiles of known drugs, allowing for faster and less costly drug development(Gazerani, 2019). Drug repositioning can also involve using an existing drug as a template for synthesizing new analogs that exhibit activity against other diseases(Cha et al., 2018). This strategy has distinct advantages over conventional drug discovery, including shorter development timelines, reduced investment needs, and higher success rates(Pushpakom et al., 2018; Wu et al., 2019). Approximately 33% of drugs approved in recent years have resulted from drug repositioning efforts, underscoring its effectiveness as a modern drug discovery strategy(Talevi& Bellera, 2020b).
Advances in computational technologies, bioinformatics, and proteomics have significantly accelerated the drug discovery process through the integration of in-silico methods(Liao et al., 2022). Computational approaches, commonly known as computer-aided drug design (CADD), have become indispensable tools at every stage of drug development(Kapetanovic, 2008). CADD enables the transformation of biological target information into computational models, allowing for data computation, analysis, and the prediction of compound activities. Molecular docking, virtual screening, and machine learning help filter large chemical libraries into smaller, more promising subsets for experimental validation. Moreover, CADD offers critical insights for lead compound optimization, focusing on improving binding affinity, pharmacokinetics, and toxicity profiles, as well as designing novel compounds via structural modifications(Sliwoski et al., 2014). The benefits of CADD are vast, providing substantial reductions in the time, cost, and experimental scope traditionally required for drug discovery. By predicting and optimizing compound properties in silico, CADD can shorten the research timeline and reduce development costs by up to 50%(Sachin S Padole et al., 2022; Xiang et al., 2012). Additionally, as computational accuracy continues to improve, these predictions increasingly align with experimental outcomes, bolstering the credibility and reliability of in silico approaches. Today, CADD is widely employed in the search for treatments for a range of diseases, including cancer(Chua et al., 2023; Reddy et al., 2007), Diabetes(Balamurugan et al., 2012; Semighini et al., 2011), and infectious diseases caused by viruses (Chen et al., 1994; Doyon et al., 2005; Yang et al., 2024)and bacteria(Duan et al., 2019; Njogu et al., 2016; Supuran, 2017).
Pyridine-4-carbohydrazide, commonly known as isoniazid, has served as a cornerstone in the treatment of tuberculosis. Its efficacy stems from its ability to inhibit the synthesis of mycolic acids, essential components of the mycobacterial cell wall(Vilchèze&Jacobs, 2019). The chemical structure of isoniazid, as depicted in Figure 1, presents a versatile molecular framework for the development of agents with a broad spectrum of biological activities.One of the common approaches isreplacing the hydrazide hydrogen with different functional groups can alter the molecule's polarity, hydrophobicity, hydrogen bonding capacity, and overall molecular conformation(Mali et al., 2021; Raczuk et al., 2022). This versatility has led to the synthesis of a plethora of derivatives with expanded therapeutic applications such as anti-inflammatory(Zhang et al., 2020), antitubercular(Aboui-Fadl et al., 2003), anticancer(Firmino et al., 2016; Rodrigues et al., 2014), antimicrobial and urease inhibitory activity(Habala et al., 2016), antidepressant and analgesic properties(Uddin et al., 2020), in the treatment of Alzheimer’s disease(Santos et al., 2020). Additionally, the pyridine moiety itself plays a crucial role in enhancing drug permeability, biochemical potency, and metabolic stability. Moreover, the pyridine ring's ability to form various non-covalent interactions with protein targets facilitates drug-target binding(Ling et al., 2021; Pennington & Moustakas, 2017). The FDA database provides compelling evidence of the prevalence of pyridine-based drugs. Approximately 18% of approved heterocyclic drugs incorporate pyridine or its derivatives, highlighting its significance as a structural motif in medicinal chemistry(Ling et al., 2021).
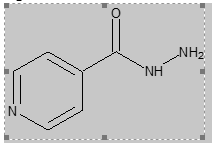
Figure 1: Chemicalstructure of Pyridine-4-Carbohydrazide (Isoniazid, INH).
Based on these advancements, this study aims to design and evaluate a chemical library of small molecules incorporating imine functional groups and pyridine-4-carbohydrazide scaffolds. Leveraging computational methods, we will predict the physicochemical properties, drug-likeness, bioactivity profiles, and ADME characteristics of these derivatives before synthesis and screening. By doing so, we aim to reduce resource waste and avoid unnecessary time and expenses associated with screening compounds that have a low likelihood of activity. This proactive approach is intended to streamline the identification of promising agents, enhancing the efficiency of the drug discovery pipeline and reducing the risk of failure in later stages.
Design Strategy
In this study, compounds (Schiff bases) were designed by utilizing the FDA-approved drug pyridine-4-carbohydrazide(A) and hybridized with substituted aldehydes (B). The imine group (C), formed during this process, was used to augment the lipophilic behavior part, as illustrated in Scheme 1. The R groups vary in type, position, partition coefficient, and hydrogen bonding capacity. This modification was intended to investigate the influence of various substituents on the phenyl ring and their impact on the biological activity of these compounds.A total of nineteen compounds were designed and assigned the code INH01-INH19.
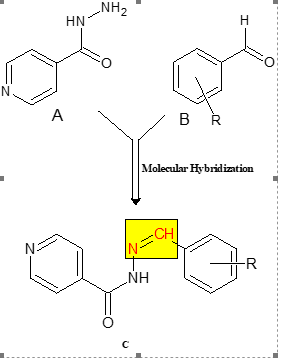
Scheme 1: The Design Concept for Schiff Base Compounds
(probably use large letter C in the figure!)
In silicoStudy
This study was conducted using a HP computer system with the following specifications: Windows 11 operating system, Intel® Core™ i5-1135G7 Quad-core Processor @ 2.40GHz, and 8 Gigabytes of RAM. The chemical structures of nineteen compounds (INH01-INH19) were initially generated using the ChemDrawsoftware suite as in Table 1A. The in-silico workflow commenced with the preparation of molecular structures for the designed compounds. These structures were first constructed in the two-dimensional (2D) format using Chem3D Ultra software and saved in the Structure Data File (SDF) format. The 2D molecular representations were then converted into SMILES (Simplified Molecular Input Line Entry System) format using the online SMILES Translator tool provided by the National Cancer Institute (https://cactus.nci.nih.gov/translate/). To ensure structural accuracy, the resulting SMILES strings were meticulously validated against their original chemical structures, as detailed in Table 1B. Following validation, the SMILES data served as the key input for subsequent predictive analysis. The data was uploaded to various online platforms, enabling the calculation of essential molecular properties and the prediction of potential biological activities for the designed compounds. This computational workflow provided valuable insights into the physicochemical, drug-likeness, bioactivity, and pharmacokinetic characteristics of the pyridine-4-carbohydrazide derivatives under investigation. The specific online platforms employed and the details of the obtained predictions are elaborated upon in subsequent sections.
Physicochemical, Drug-likeness, and bioactivity Properties predictions
The physicochemical properties, drug-likeness, and bioactivity of the compounds were evaluated using the molinspirationchemoinformatics platform. Drug-likeness assessments were conducted according to Lipinski's Rule of Five and Veber's rules. Additionally, the platform predicted bioactivity against six different protein targets: GPCR, ICM, KI, NRC, PI, and EI.
Prediction of Activity Spectra for Substances (PASS)
To further elucidate the biological activity spectrum of the pyridine-4-carbohydrazide derivatives, the PASS online tool (https://www.way2drug.com/passonline/) was employed.
ADME Prediction
The ADME properties of the derivatives were predicted using the pkCSM-Pharmacokinetics software (https://biosig.lab.uq.edu.au/pkcsm/). Established methodologies documented in the scientific literature served as the foundation for these predictions.
Table 1A: Codes and chemical Structures of the designed compounds.
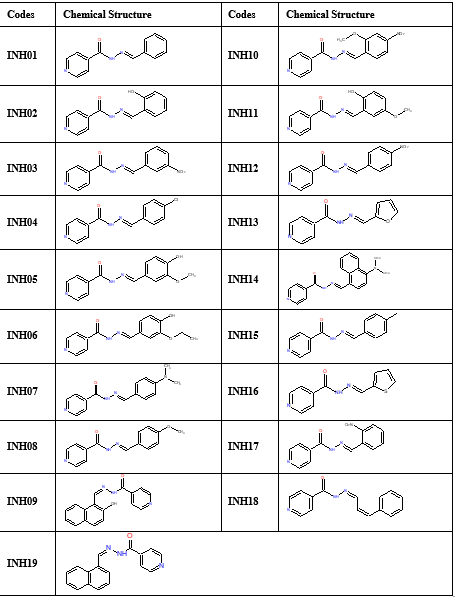
Table 1B: IUPAC names and canonical SMILES of the predicted compounds.
| Codes | Character | |
| INH01 | IUPAC name | N'-[phenylmethylidene] pyridine-4-carbohydrazide |
| SMILES | O=C(NN=Cc1ccccc1)c2ccncc2 | |
| INH02 | IUPAC name | N-(2-hydroxybenzylidene) pyridine-4-carbohydrazide |
| SMILES | O=C(NN=Cc1ccccc1O)c2ccncc2 | |
| INH03 | IUPAC name | N'-[(3-nitrophenyl) methylidene] pyridine-4-carbohydrazide |
| SMILES | C1=CC(=CC(=C1)[N+](=O)[O-])C=NNC(=O)C2=CC=NC=C2 | |
| INH04 | IUPAC name | N`-[(Z)-(4-Chlorophenyl) methylidene] pyridine-4-carbohydrazide |
| SMILES | ClC1=CC=C(\C=N\NC(=O)C2=CC=NC=C2)C=C1 | |
| INH05 | IUPAC name | N-[(4-Hydroxy-3-methoxybenzylidene) pyridine-4-carbohydrazide |
| SMILES | COC1=C(C=CC(=C1)C=NNC(=O)C2=CC=NC=C2)O | |
| INH06 | IUPAC name | (N'-(3-ethoxy-4-hydroxybenzylidene) pyridine-4-carbohydrazide |
| SMILES | CCOc2cc(C=NNC(=O)c1ccncc1)ccc2O | |
| INH07 | IUPAC name | N-(4-(dimethylamino) benzylidene) pyridine-4-carbohydrazide |
| SMILES | CN(C)C1=CC=C(C=C1)C=NNC(=O)C2=CC=NC=C2 | |
| INH08 | IUPAC name | (E)-N'-(4-methoxybenzylidene) pyridine-4-carbohydrazide |
| SMILES | COc2ccc(C=NNC(=O)c1ccncc1)cc2 | |
| INH09 | IUPAC name | (E)-N'-((2-hydroxynaphthalen-1-yl) methylene) pyridine-4-carbohydrazide |
| SMILES | O=C(NN=Cc1c(O)ccc2ccccc12)c3ccncc3 | |
| INH10 | IUPAC name | N'-(2-methoxy-4-nitrobenzylidene) pyridine-4-carbohydrazide |
| SMILES | COc1cc(N(=O)=O)ccc1C=NNC(=O)c2ccncc2 | |
| INH11 | IUPAC name | N'-(2-hydroxy-5-methoxybenzylidene) pyridine-4-carbohydrazide |
| SMILES | COc2ccc(O)c(C=NNC(=O)c1ccncc1)c2 | |
| INH12 | IUPAC name | N'-(4-nitrobenzylidene) pyridine-4-carbohydrazide |
| SMILES | O=C(NN=Cc1ccc(N(=O)=O)cc1)c2ccncc2 | |
| INH13 | IUPAC name | N'-(furan-2-ylmethylene)pyridine-4-carbohydrazide |
| SMILES | O=C(NN=Cc1ccco1)c2ccncc2 | |
| INH14 | IUPAC name | N'-((4-(dimethylamino) naphthalen-1-yl) methylene) pyridine-4-carbohydrazide |
| SMILES | CN(C)c2ccc(C=NNC(=O)c1ccncc1)c3ccccc23 | |
| INH15 | IUPAC name | N-(4-methylbenzylidene) pyridine-4-carbohydrazide |
| SMILES | CC1=CC=C(C=C1)C=NNC(=O)C2=CC=NC=C2 | |
| INH16 | IUPAC name | N'-[thiophen-2-yl] pyridine-4-carbohydrazide |
| SMILES | C1=CSC(=C1)/C=N\NC(=O)C2=CC=NC=C2 | |
| INH17 | IUPAC name | N'-[(2-nitrophenyl) methylidene] pyridine-4-carbohydrazide |
| SMILES | C1=CC=C(C(=C1)/C=N/NC(=O)C2=CC=NC=C2)[N+](=O)[O-] | |
| INH18 | IUPAC name | N-[(E)-[(Z)-3-phenylprop-2-enylidene]amino] pyridine-4-carbohydrazide |
| SMILES | C1=CC=C(C=C1)/C=C\C=N\NC(=O)C2=CC=NC=C2 | |
| INH19 | IUPAC name | N'-[(E)-(naphthalen-2-yl) methylidene] pyridine-4-carbohydrazide |
| SMILES | O=C(NN=Cc1cccc2ccccc12)c3ccncc3 | |
| INH | IUPAC name | Pyridine-4-carbohydrazide |
| SMILES | C1=CN=CC=C1C(=O)NN |
Table 1C: Distribution of predictors used in the in-Silico Study.
| Parameter | Predictor | Unit | Requirement value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Physiochemical and drug-likeness properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Lipinski's Rule
| Molecular weight (MW) | g/mol | <500> | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Partition coefficient (LogP) | - | < 5> | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of hydrogen bond acceptors | - | <10> | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of hydrogen bond donors | - | < 5> | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Veber Rule | Number of rotatable bonds (N-rotb) | - | < 5> | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Topological polar surface area (TPSA) | - | <140> | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bioactivity score properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bioactivityscore | G-protein-coupledreceptor ligands (GPCR) | - |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ion channel modulation (ICM) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Kinase inhibitor (KI) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Nuclear receptor ligands (NRL) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Protease inhibitor (PI) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enzyme inhibitor (EI) | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pharmacokinetic properties | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Absorption | Water solubility | LogS | - | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Intestinal absorption (HIA) |
Results and discussionDesign strategy This study presents an approach to the repositioning of isoniazid by introducing structural modifications at the terminal at the terminal NH2 group. The strategy involves hybridizing isoniazid with various aromatic or heteroaromatic aldehydes,leading to the formation of a new functional group known as an imine. The selection of aromatic aldehydes as hybridization partners was motivated by their structural features. Compared to chains, aromatic aldehydes exhibit fewer degrees of freedom, which can contribute to enhanced ligand-receptor binding energy by reducing the entropic penalty. This property can potentially lead to increased compound potency(Mushtaque & Rizvi, 2023; Rohilla et al., 2024). The designed compounds possess several key features:
The structural modifications introduced in these compounds are expected to significantly influence their physicochemical properties, including lipophilicity, electronic characteristics, and steric effects. These alterations may, in turn, lead to changes in biological activity and therapeutic potential. Ultimately, the goal of these modifications is to improve the compounds' efficacy, particularly for applications as DNA-binding agents. Physiochemical and drug-likeness properties predictions Prediction of the physicochemical properties of drugcandidates is essential for efficient drug development and understanding their biological and medicinal actions(Leeson & Young, 2015; Meanwell, 2011). Properties such as molecular weight, the number of rotatable bonds, and the number of heavy atoms are integral to evaluating drug-likeness, helping identify oral drug candidates in the early phases of drug discovery(Lee et al., 2022a; Tripathi &Ayyannan, 2018).Drug-like compounds are molecules that contain functional groups and/or have physical properties consistent with the majority of known drugs, suggesting that these compounds could potentially exhibit biological activity or therapeutic effects. The drug-like characteristics serve as a parameter in choosing a more promising compound as a lead from the extensive combinatorial libraries(Lee et al., 2022b; Tian et al., 2015). One of the foundational methods for assessing drug-like properties is Lipinski’s Rule of Five (Ro5), developed by Pfizer's medicinal chemist, Christopher Lipinski. The RO5 was derived from an analysis of orally available drugs and clinical candidates, though it excludes certain classes such as antibiotics, antifungals, vitamins, and cardiac glycosides. The RO5 states that a compound is more likely to be membrane permeable and easily absorbed via passive diffusion in human intestine if it meets the following criteria: molecular weight (MW) <500> Molinspiration web-based software plays a pivotal role in evaluating physicochemical properties and drug-likeness. This tool leverages advanced Bayesian statistical methods, integrating the structural and property data of both active and inactive compounds to identify substructural features characteristic of biologically active molecules. The software calculates key physicochemical parameters crucial for predicting the theoretical oral bioavailability of the compounds under investigation. These parameters include molecular weight, partition coefficient (logP), the number of hydrogen bond acceptors and donors, the number of rotatable bonds, and total polar surface area, as outlined in Table 4A. Number of heavy atoms (N atoms): The number of heavy atoms in a molecule is a key factor in drug design, influencing molecular size, complexity, drug-likeness, and pharmacokinetic properties(García-Sosa et al., 2012). While larger molecules offer increased binding potential, they may also pose challenges related to synthesis, solubility, and membrane permeability. During lead optimization, reducing the number of heavy atoms can lower molecular weight and improve solubility, enhancing the drug-likeness profile without significantly affecting potency(de Souza Neto et al., 2020; Wang et al., 2019). Isoniazid derivatives contain between 16 and 24 heavy atoms, with INH having the lowest count at 10 atoms. The increase in heavy atoms in derivatives is often due to the substitution of larger aromatic rings or additional functional groups, such as in INH14, which has the highest count due to the inclusion of a naphthalene ring and a dimethylamino group. Molecular weight (MW): Molecular weightis the sum of the atomic weights of all atoms in a molecule, typically expressed in Daltons (Da) or grams per mole (g/mol). MW is a critical determinant of a drug’s absorption, distribution, metabolism, and excretion (ADME) profile(Komura et al., 2023). As MW increases, drug permeability and absorption generally decrease, particularly concerning membrane permeability and penetration through the blood-brain barrier (BBB)(Pardridge, 2012). Additionally, drug clearance through artificial membranes inversely correlates with MW(Lienx& Wang, 1980). The MW values of the designed compounds range from 215.21 to 318.38 Da, all of which are under the 500 Da threshold, suggesting that these molecules are likely to be easily absorbed and exhibit good permeability across cell membranes. Partition coefficient (LogP): The partition coefficient is a key measure of lipophilicity or hydrophobicity, calculated as the logarithm of the concentration ratio of a compound between organic (usually n-octanol) and aqueous phases(Ruiz-Garcia et al., 2008). Positive values of the partition coefficient suggest a tendency towards a lipophilic or hydrophobic environment,whereas negative values indicate a preference for a lipophobic or hydrophilic environment(Khanna & Ranganathan, 2009). LogP values significantly impact various ADMET parameters, drug-receptor interactions, and the overall potency of molecules(Tshepelevitsh et al., 2020). Compounds exhibiting extremely high or low LogP values may encounter challenges related to permeability and solubility(Waring, 2010). Highly hydrophilic compounds generally struggle to diffuse passively through cellular membranes due to their inability to penetrate the hydrophobic core of the lipid bilayer. Conversely, excessively lipophilic compounds may also face difficulties in membrane permeation, as they tend to become sequestered within the bilayer, impeding their effective transit(Lagorce et al., 2017).Researchers have also identified a correlation between a compound's logP value and its ability to penetrate the blood-brain barrier (BBB), a crucial factor for central nervous system (CNS) activity. For CNS-active drugs, a logP value in the range of 4 to 5 is generally considered optimal(Abraham et al., 1993; Feher et al., 2000). The LogP values for the designed compounds (INH01–INH19) were within the acceptable range per Lipinski’s Rule and showed superior lipophilicity compared to the precursor compound, isoniazid (INH), which had a negative LogP value. This enhancement in lipophilicity, likely attributed to the presence of the Imine group and aromatic portion of the aldehyde, suggests enhanced absorption through biological membranes. Hydrogen bond acceptor and hydrogen bond donor groups: Hydrogen bonds play a crucial role in molecular recognition(Morozov &Kortemme, 2005; Santos-Martins & Forli, 2020), structural stability(Pace et al., 2011), enzyme catalysis(Calixto et al., 2019; Neves et al., 2017), and drug partition and permeability(Rezai et al., 2006; Shinoda, 2016). The presence of functional groups capable of forming hydrogen bonds can enhance a drug's solubility and its ability to interact with biomolecular targets, thereby influencing binding affinity and selectivity. However, an excess of hydrogen bond donors or acceptors can negatively impact membrane permeability and partitioning(Alex et al., 2011). In that regard, drug-like character predictors, such as Lipinski's rule of five (Ro5) have been using the number of hydrogen bond donors/acceptors as a molecular descriptor.In the designed compounds, the number of HBAs ranges from 4 to 8, and the number of HBDs ranges from 1 to 2, all within the limits set by the Ro5, ensuring that the hydrogen bonding potential does not compromise the compounds' drug-like properties. The number of rotatable bonds (N rotb): The number of rotatable bonds is a measure of molecular flexibility and is an important descriptor for predicting oral bioavailability. A rotatable bond is defined as any single bond not part of a ring and bound to a non-terminal heavy atom(Veber et al., 2002). A higher number of rotatable bonds increases molecular flexibility, potentially improving binding affinity with target proteins. However, compounds with fewer rotatable bonds are generally more rigid, which can enhance oral bioavailability by reducing the entropy cost of binding(Vieth et al., 2004). In the designed compounds, the number of rotatable bonds does not exceed 10, which is favorable for maintaining good oral bioavailability. Topological polar surface area (TPSA): Topological polar surface area is a key descriptor related to hydrogen bonding and is important for drug transport properties such as intestinal absorption, BBB penetration, and oral bioavailability(Leeson, 2016; Veber et al., 2002). TPSA is calculated as the sum of the surface areas of polar atoms, primarily oxygen, and nitrogen, including attached hydrogens(Ertl et al., 2000). Therefore, TPSA is a reliable indicator of a compound's hydrogen bonding capacity. Compounds with a TPSA below 140 Ų typically exhibit good permeability and oral absorption, while those with a TPSA below 80 Ų are more likely to permeate the CNS by passive diffusion(Clark, 2011). The TPSA values for the isoniazid derivatives range from 54.59 to 109.14 Ų, well below the 140 Ų threshold, suggesting favorable absorption characteristics. Molecular volume (Å3): Molecular volume, a fundamental property of a molecule, plays a pivotal role in drug discovery. Its significance extends to optimizing drug candidates, understanding interactions with biological targets, and predicting pharmacokinetic properties(Flatow et al., 2014). By determining the molecular volume of a compound, researchers can gain insights into its binding affinity, solubility, permeability, and overall drug-likeness(La-Scalea et al., 2005; Mcgowan, 1956). In the context of isoniazid derivatives, molecular volume variations offer valuable information. The range of molecular volumes observed in these derivatives, from 187.51 ų to 295.84 ų, highlights the influence of structural modifications on the overall molecular size and shape. This understanding can inform the design of novel derivatives with improved properties. Bioactivity predictions In drug discovery, predicting the bioactivity of compounds against specific biological targets is crucial for understanding their potential therapeutic effects and toxicity profiles. Biological targets, which commonly include proteins,whether cytosolic, or membrane-embedded,and nucleic acids, play a key role in mediating the biological activity of drugs(Decherchi& Cavalli, 2020). To assess the likelihood of compounds interacting with these targets, bioactivity scores can be calculated using tools like Molinspiration, an open-source chemoinformatics platform. These scores provide valuable insights into compounds' binding affinity and selectivity, facilitating the development of new drugs with enhanced efficacy and reduced side effects.Bioactivity scores are categorized as follows: compounds with scores greater than 0.0 are likely to exhibit significant biological activity, scores between -0.50 and 0.00 suggest moderate activity, and scores below -0.50 indicate inactivity(Khan et al., 2017). Table 4B suggests the compounds exhibit moderate to inactive interactions with six protein targets. The efficiency of bioactivity scores typically follows the order of Enzyme Inhibitors (EI), Kinase Inhibitors (KI), G-protein-coupled receptors (GPCR), Protease Inhibitors (PI), Nuclear Receptors (NRC), and Ion Channel Modulators (ICM). The bioactivity profiles of isoniazid derivatives (INH01 to INH19) vary significantly depending on the structural modifications made to the pyridine-4-carbohydrazide core. For instance, the parent compound, isoniazid, shows relatively low bioactivity scores across all six targets, particularly in the categories of Nuclear Receptors (NRC: -2.33) and Ion Channel Modulators (ICM: -1.45). This suggests a low predicted affinity for these targets, consistent with isoniazid's primary role as an antibiotic rather than a modulator of these pathways. Derivatives such as INH01 (Phenylmethylidene) exhibit moderate activity across all targets, indicating that simple phenyl substitution does not dramatically alter activity compared to the parent compound. In contrast, derivatives like INH13 (Furan-2-yl) and INH16 (Thiophen-2-yl), which feature heterocyclic aromatic rings, display the lowest activity scores, implying that these substitutions may lead to reduced bioactivity. The presence of sulfur in the thiophene ring of INH16 could contribute to an electron-rich environment that is less favorable for target interactions. Compounds with strong electron-withdrawing groups, such as INH03 (3-Nitrophenyl) and INH17 (2-Nitrophenyl), show decreased bioactivity across all targets, particularly for ICM and NRC. This suggests that the electron-withdrawing nature of the nitro group reduces binding affinity. Conversely, electron-donating groups like methoxy and hydroxy in derivatives such as INH05 (4-Hydroxy-3-methoxyphenyl) and INH11 (2-Hydroxy-5-methoxyphenyl) slightly improve bioactivity, possibly by enhancing electronic interactions with certain targets. Derivatives with dimethylamino groups, such as INH07 (4-Dimethylaminobenzylidene) and INH14 (4-(Dimethylamino)naphthalen-1-yl), show moderate activity, indicating that electron-donating groups can enhance bioactivity, though the overall effect also depends on the position and presence of additional substituents. Meanwhile, bulky naphthalene-containing derivatives like INH09 (2-Hydroxynaphthalen-1-yl) and INH19 (Naphthalen-2-yl) generally exhibit low bioactivity, likely due to increased steric hindrance that reduces effective target binding. Lastly, INH18 ((Z)-3-phenylprop-2-enylidene) shows moderate bioactivity, suggesting that some flexibility in molecular structure can slightly improve target binding, though not significantly. PASS prediction The Prediction of Activity Spectra for Substances (PASS) platform provides a robust computational approach for forecasting the biological activity spectra of chemical compounds. Developed by the V. N. Orechovich Institute of Biomedical Chemistry, PASS predicts pharmacological effects based on structural similarities to known biologically active compounds. This model relies on a vast dataset, primarily derived from the MDL Drug Data Report (MDDR), and continuously updated to reflect discoveries in medicinal chemistry(Parasuraman, 2011). The interpretation of PASS predictions requires a degree of flexibility, particularly concerning the values of Pa. A Pa value greater than 0.7 indicates a strong probability that the predicted biological activity can be experimentally confirmed. This suggests that the compound shares significant structural similarities with known pharmacologically active agents, making it a promising candidate for experimental validation. For Pa values between 0.5 and 0.7, the probability of experimental confirmation is lower. However, such compounds may exhibit novel structural features that are not closely aligned with known drugs. These novel features could offer valuable insights into unique or less common mechanisms of action. In contrast, a P value below 0.5 suggests a relatively low probability of experimental validation. Nonetheless, compounds with low Pa values may still exhibit structural novelty, presenting opportunities for discovery in previously unexplored areas of biological activity(Filimonov et al., 2014). Table 5 presents the PASS predictions for a series of pyridine-4-carbohydrazide derivatives (INH01–INH19), focusing on the most promising activities where Pa > 0.7. These predictions aim to understand the potential therapeutic applications of these compounds, with isoniazid (INH) serving as the template compound.Based on the data, each compound has multiple potential activities, we can classify the activities into several categories: Core Activities: These activities appear consistently across multiple INH compounds and likely represent the primary mechanisms of action: antituberculosis, antimycobacterial, taurine dehydrogenase inhibitor, and amine dehydrogenase inhibitor. Secondary Activities: These are activities that appear in multiple INH compounds but with less frequency or probability than the core activities. HMGCS2 expression enhancer (cholesterol metabolism), phosphatidylserine decarboxylase inhibitor (cell signaling), glutamine-phenylpyruvate transaminase inhibitor (amino acid metabolism), antiviral (picornavirus and poxvirus), beta-adrenergic receptor kinase inhibitor (hormonal signaling). Tertiary Activities: These are less common and often have lower probabilities. Threonine aldolase inhibitor, isopenicillin-N epimerase inhibitor, nicotinamidase inhibitor, PFA-M1 aminopeptidase inhibitor, MCL-1 antagonist,nicotinic alpha6beta3beta4alpha5 receptor antagonist, corticosteroid side-chain-isomerase inhibitor, phenylalanine(histidine) transaminase inhibitor, CYP2A8 substrate, gluconate 2-dehydrogenase (acceptor) inhibitor, cytoprotectant, transcription factor stat3 inhibitor, arylalkyl acylamidase inhibitor, aldehyde dehydrogenase (pyrroloquinoline-quinone) inhibitor, thiol protease inhibitor, neuropeptide y2 antagonist, aspartate-phenylpyruvate transaminase inhibitor, phthalate 4,5-dioxygenase inhibitor, glycosylphosphatidylinositol phospholipase D inhibitor. Additionally, the analysis of predicted activities across the INH derivatives reveals some interesting structure-activity relationships (SARs). Antibacterial activity, a primary focus due to INH’s established efficacy against Mycobacterium tuberculosis and Gram-positive bacteria, showed that many derivatives had comparable or even superior potency to INH. Structural modifications appear to enhance antibacterial potency, especially with para-substitution on the phenyl ring, which was particularly effective for antimycobacterial activity. Derivatives such as INH04, INH07, INH08, INH12, and INH15 exhibited higher activity with para-substituted phenyl groups. Electron-donating groups (e.g., -OCH3, -N(CH3)2) further improved activity, as seen in INH07, INH08, and INH14, while electron-withdrawing groups (e.g., -NO2, -Cl) also boosted potency, as observed in INH03 and INH12. Compounds containing furan (INH13) or thiophene (INH16) substituents showed strong antibacterial activity, particularly against tuberculosis. In contrast, ortho- or meta-substitution (INH02, INH05, INH06, INH11) tended to reduce activity compared to para-substituted compounds. Naphthyl substitutions at the 1- or 2-position (INH09, INH14, INH19) did not significantly enhance activity over phenyl derivatives, and the lack of activity in INH14 suggests that bulkier groups may interfere with key binding interactions. Regarding antiviral activity, isoniazid itself demonstrated no potential across viral targets. However, many derivatives exhibited enhanced antiviral properties, underscoring the importance of structural modifications. Derivatives such as INH01, INH03, INH04, INH07, INH08, INH10, INH12, INH15, INH17, and INH19 displayed notable antiviral activity, with INH01, INH07, INH08, INH12, and INH15 showing efficacy against Picornavirus (PV) and Poxvirus (POV). Similar to the antibacterial SARs, para-substitution on the phenyl ring was again favored for antiviral activity. Electron-donating groups (e.g., -OCH3, -N(CH3)2) boosted activity in compounds like INH07, INH08, and INH14, while electron-withdrawing groups (e.g., -NO2, -Cl) enhanced activity in INH03 and INH12. Furan (INH13) and thiophene (INH16) substituents showed little antiviral activity, while ortho- or meta-substituted phenyl groups (INH02, INH05, INH06, INH11) performed less effectively. Naphthyl substituents (INH09, INH14, INH19) were not advantageous compared to phenyl groups. INH04, INH07, INH08, INH12, and INH15 emerge as promising candidates for further investigation, warranting in vitro and in vivo studies to confirm their antiviral efficacy and safety. In contrast, INH demonstrated no predicted activity as an enhancer of HMGCS2 expression, a key enzyme in lipid metabolism. However, many derivatives showed increased activity in this area, indicating that structural modifications improved the HMGCS2 expression-enhancing properties. Para-substitution on the phenyl ring was again advantageous for enhancing HMGCS2 expression. Compounds such as INH04, INH07, INH08, INH12, and INH15 showed heightened activity, with electron-donating groups (e.g., -OCH3, -N(CH3)2) further enhancing efficacy, as in INH07, INH08, and INH14. Electron-withdrawing groups (e.g., -NO2, -Cl) also proved beneficial, as seen in INH03 and INH12. INH13, containing a furan substituent, exhibited the highest activity for HMGCS2 expression enhancement, underscoring the importance of this group. As with antibacterial and antiviral activities, ortho- and meta-substitutions (INH02, INH05, INH06, INH11) resulted in reduced activity, while naphthyl substituents (INH09, INH14, INH19) did not significantly improve activity over phenyl groups. Derivatives INH02, INH10, INH12, and INH13 are particularly promising for further exploration in metabolic or transcriptional pathways regulated by HMGCS2. ADME prediction As opposed to pharmacodynamics, which describes what the drug does to the body. Pharmacokinetics (PK) describes what the body does to the drug(Currie, 2018a). There are four major determinants of PK, commonly called ADME properties(absorption, distribution, metabolism, and excretion). These properties are crucial for determining the drug's efficacy and safety(Lucas et al., 2019). The ADME process can be broadly summarized as (i) drug dissolution in the gastrointestinal tract, followed by absorption through the gut wall and passage into the bloodstream via the liver; (ii) distribution of the drug to various tissues, depending on its structural and physicochemical properties; (iii) metabolism, where the drug is biochemically modified into metabolites, often by enzymatic systems, and (iv) elimination of the drug, usually through excretion (Gleeson et al., 2011; van de Waterbeemd& Gifford, 2003). For a compound to be effective it must reach its target in the body at sufficient concentrations and remain in a bioactive form long enough to exert its intended biological effects. Thus, understanding ADME properties early in drug development can help minimize the time, cost, and labor involved by focusing on compounds with promising profiles. This section evaluates the absorption, distribution, metabolism, and excretion characteristics of pyridine-4-carbohydrazide derivatives (INH01–INH19), comparing them to the parent compound isoniazid (INH). (A) Absorption Absorption refers to the process by which a drug moves from the site of administration into systemic circulation(Currie, 2018b). Several parameters are used to evaluate the absorption potential of drug candidates: water solubility (LogS), human intestinal absorption (HIA), permeability across the Caco-2 cell line (LogPapp), skin permeability (LogKp), and their interactions with P-glycoprotein (P-gp I, II). These factors influence the bioavailability of a compound, especially when administered orally. The results of these parameters are summarized in Table 6A. Water solubility (logS):Water solubility is critical for drug formulation and absorption, particularly for oral delivery(Barrett et al., 2022;Delaney, 2004). Low solubility can lead to poor bioavailability and impaired absorption, while high solubility can enhance drug dissolution and plasma concentration(Tran et al., 2023). The aqueous solubility of a substance is often expressed as log units of molar solubility (mol/L), or logS. The pyridine-4-carbohydrazide derivatives exhibit varying degrees of water solubility, ranging from -1.8896 to -4.042. Negative values reflect lower solubility. For example, INH01 (-2.076) and INH02 (-2.894) exhibit better solubility than isoniazid (-1.6), possibly due to simpler aromatic substitutions like phenyl (INH01) or hydroxyl groups (INH02), which slightly decrease solubility without significantly impairing absorption. On the other hand, derivatives like INH10 (-4.02), INH14 (-4.042), and INH17 (-3.643) show poor solubility, likely because of bulky or electron-withdrawing groups, such as nitro or naphthyl rings, which hinder aqueous solubility. These low solubility values suggest that these derivatives may struggle with absorption, particularly in aqueous environments. Human intestinal absorption (HIA):HIA reflects how well a compound is absorbed through the intestinal lining(Azman et al., 2022), and a value over 80% is considered indicative of good absorption(Chander et al., 2017; Pires et al., 2015). The HIA values for the pyridine-4-carbohydrazide derivatives range from 83.21% to 96.43%, indicating that all compounds have good predicted absorption and are favorable for oral bioavailability. INH05 (96.317%) and INH14 (96.436%) show the highest absorption rates, suggesting that their methoxy-hydroxyphenyl and dimethylamino-naphthyl groups, respectively, enhance lipophilicity and passive diffusion through cell membranes. Conversely, INH10 (83.223%) and INH03 (85.889%) demonstrate lower absorption due to the presence of nitro groups, which tend to reduce membrane permeability. Isoniazid itself shows a lower HIA value (75.651%), likely due to its simple structure and lack of lipophilic substituents that facilitate passive absorption. The Caco-2 cell line (Caco-2):The Caco-2 cell line is often used as a model for intestinal permeability(Kus et al., 2023). Compounds with a LogPapp value greater than 0.90 cm/s are considered to have high permeability(Pires et al., 2015). The pyridine-4-carbohydrazide derivatives show varying permeability values, ranging from -0.1 to 1.386. Compounds such as INH04, INH06, INH07, and INH09 exhibit high permeability, while others, including INH02, INH03, INH10, INH12, INH13, INH14, and INH17, show lower permeability. The reduced permeability of these compounds may be attributed to the presence of polar, bulky, or electron-withdrawing groups, which can impede passive diffusion across intestinal membranes. Skin permeability (LogKp):Skin permeability reflects the ability of a drug to penetrate the skin barrier, a critical factor for transdermal drug delivery systems(Cordery et al., 2017; Pensado et al., 2022; Tsakovska et al., 2017). Compounds with a LogKp value greater than -2.5 cm/h are considered to have relatively low skin permeability(Pires et al., 2015). The LogKp values of the pyridine-4-carbohydrazide derivatives range from -3.29 to -2.398, suggesting limited potential for transdermal absorption in most compounds. However, INH18, with a LogKp value of -2.398, exhibits the highest skin permeability among the derivatives, indicating that this compound may have potential for transdermal drug delivery. Permeability glycoprotein (P-gp) interaction:P-gp is a membrane-bound efflux transporter that can limit the bioavailability of drugs by actively pumping them out of cells, particularly in tissues such as the intestines, liver, and brain(Saaby & Brodin, 2017). Compounds that are substrates for P-gp may face reduced bioavailability, as the transporter pumps them out of cells before they can reach therapeutic concentrations(Elmeliegy et al., 2020; Nielsen et al., 2023). Moreover, P-gp substrates can be further categorized into drugs that are not metabolized in humans and those that are substrates for both P-gp and drug-metabolizing enzymes, particularly CYP3A4. Given that many P-gp substrates are also metabolized by CYP3A4, and that P-gp inhibitors often inhibit CYP3A4 as well, numerous drug-drug interactions arise from the inhibition or induction of both P-gp and CYP3A4(Fromm, 2004; König et al., 2013). Several compounds in the study, including INH03, INH07, INH08, INH09, and INH12, are predicted to be substrates for P-gp, suggesting they may face reduced bioavailability due to efflux and possible metabolism by CYP3A4. In contrast, isoniazid and the other derivatives are not expected to interact with P-gp, which may result in better bioavailability by avoiding efflux. Additionally, most compounds are not predicted to inhibit P-gp, except for INH14, which inhibits both P-gp I and II. This inhibition could enhance bioavailability by preventing the efflux of co-administered drugs, making INH14 a potential candidate for combination therapies aimed at overcoming multidrug resistance, particularly in cancer treatments(Côrte-Real et al., 2019; Dong et al., 2020; Waghray & Zhang, 2018).
(D) Distribution Drug distribution refers to the reversible transfer of a drug within the body, from the bloodstream to various tissues(Berellini et al., 2009; Motl et al., 2006). It plays a crucial role in the ADMET process, as it influences the amount of drug that reaches target sites, affecting both efficacy and potential toxicity(Sun et al., 2022). The distribution properties of derivatives are evaluated using four key parameters: volume of distribution, fraction unbound, blood-brain barrier permeability, and central nervous system permeability, as shown in Table 6B. The Volume of Distribution (VDss) quantifies how extensively a drug disperses into body tissues relative to the bloodstream. Higher VDss values (closer to positive) indicate a greater extent of tissue distribution(Hsu et al., 2021). According to Pires et al., a compound is considered to have good tissue distribution if its VDss value exceeds 2.81 L/kg (log VDss> 0.45) and poor distribution if it is below 0.71 L/kg (log VDss< -0.15)(Pires et al., 2015). The VDss values for the derivatives range from -0.432 to 0.212 Log L/kg, suggesting low to moderate tissue distribution. Notably, compounds such as INH02, INH07, INH08, INH09, INH14, INH15, and INH19 exhibit moderate tissue distribution, likely due to their aromatic structures, which enhance lipophilicity compared to isoniazid and other derivatives. Fraction Unbound (FU) represents the proportion of a drug in the plasma that remains unbound to proteins, with only the unbound fraction being pharmacologically active(Seyfinejad et al., 2021). A higher FU indicates a greater portion of the drug available to exert therapeutic effects(Watanabe et al., 2018). The FU values for the derivatives range from 0.031 to 0.333, with lower FU values corresponding to higher protein binding. Compounds such as INH14, INH19, and INH09, which contain extensive aromatic substituents, and INH03, INH10, and INH17, which contain nitro groups, show lower FU values. In contrast, isoniazid demonstrates the highest FU (0.728), significantly greater than most derivatives, reflecting minimal protein binding and suggesting a higher bioavailability for interaction with target sites. Blood-Brain Barrier (BBB) Permeability indicates a drug’s ability to cross the BBB, a selective barrier that regulates the entry of substances into the brainCrivori et al., 2000). Compounds with a LogBB> 0.3 are considered capable of crossing the BBB easily, while those with a LogBB< -1.0 face significant barriers(Pires et al., 2015). Most pyridine-4-carbohydrazide derivatives demonstrate low BBB permeability, suggesting limited brain penetration. However, compounds INH15 and INH18 exhibit higher BBB permeability, indicating their potential for greater brain access. CNS Permeability (Log PS) further evaluates the ability of these compounds to penetrate the central nervous system. Compounds with a Log PS > -2 are considered capable of CNS penetration, while those with a Log PS < -3 are unlikely to cross the CNS barrier(Pires et al., 2015). For the derivatives studied, only INH19 exhibits a Log PS lower than -2, suggesting it has poor CNS penetration. The remaining derivatives exhibit moderate CNS permeability (Log PS between -2 and -3). Isoniazid, with a Log PS of -3.022, shows one of the lowest CNS permeabilities, indicating it is less likely to penetrate the CNS effectively, which aligns with its hydrophilic structure. 3.5.3 | (M) Metabolism Drug metabolism is the process by which the body's enzymes chemically modify drug molecules. This is a vital defense mechanism against potential toxins, which are often lipid-soluble and can accumulate in the body. To facilitate excretion, these toxins are converted into more water-soluble metabolites.Most drug metabolism occurs in the liver, where enzymes called hepatic microsomal enzymes catalyze the breakdown process.The metabolic properties of pyridine-4-carbohydrazide derivatives were assessed by evaluating their potential as substrates and/or inhibitors of key cytochrome P450 (CYP) enzymes, which are essential detoxifying enzymes predominantly expressed in the liver(Zanger & Schwab, 2013). To date, 57 distinct CYP isoforms have been identified in humans, of which five—CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4—play pivotal roles in drug metabolism(Wei et al., 2024). The metabolic pharmacokinetic characteristics of the compounds are presented in Table 6C. Among these isoforms, CYP3A4 and CYP2D6 are of particular clinical importance due to their significant involvement in the metabolism of various drugs. Inhibition of these enzymes can result in reduced drug clearance, elevated drug plasma concentrations, and potential adverse reactions. Conversely, if a compound serves as a substrate for these enzymes, it is likely to be efficiently metabolized, reducing the risk of side effects related to drug accumulation.Several pyridine-4-carbohydrazide derivatives, including INH01, INH03, INH04, INH07, INH08, and INH10, are identified as substrates for CYP3A4, indicating their likelihood of efficient metabolic processing by this enzyme, thereby reducing the risk of accumulation-related side effects. Furthermore, most derivatives exhibit no significant inhibition of CYP2D6, CYP2E1 and CYP3A4, which is advantageous, as it suggests a lower potential for drug-drug interactions and reduced risk of hepatotoxicity. Notably, INH18 acts as a dual substrate for both CYP3A4 and CYP2D6, further lowering the probability of metabolic interactions. Regarding CYP1A2, all derivatives except INH06 inhibit this isoform, potentially resulting in elevated plasma concentrations of co-administered drugs metabolized by CYP1A2 and increasing the risk of drug-drug interactions. The inhibition of CYP1A2 may be linked to the presence of electron-withdrawing substituents, such as nitro and halogen groups, on the aromatic rings of these compounds, which likely facilitate interaction with the enzyme's active site. About CYP2C8, most derivatives inhibit this enzyme, with the exceptions of INH07, INH12, INH13, INH15 and INH16, which do not exhibit inhibitory activity. The inhibition observed in other derivatives may be attributed to structural features, such as bulky substituents, which could impede the enzyme's binding affinity. For CYP2C9, only INH14 shows inhibitory activity, which may be ascribed to the presence of a dimethylaminonaphthyl group that interacts with the active site of CYP2C9. Several derivatives also exhibit inhibition of CYP2C19, including INH04, INH09, INH10, INH14, INH18, and INH19. Structural elements such as nitro groups in INH09 and INH10, and naphthyl groups in INH14 and INH19, likely contribute to this inhibitory activity by introducing steric and electronic effects that influence binding to the CYP2C19 active site. (E) Excretion Excretion refers to the process by which drugs are eliminated from the body, and it is closely related to the concentration of the drug in the bloodstream and the rate of elimination(Talevi& Bellera, 2018). In this study, the excretion properties of pyridine-4-carbohydrazide derivatives (INH01–INH19) were evaluated based on their total clearance (CLtot) values, expressed in Log ml/min/kg, and their potential interaction with the Renal Organic Cation Transporter 2 (OCT2), as presented in Table 6D. The total clearance (CLtot) values for the derivatives ranged from 0.027 to 0.966 Log ml/min/kg. Isoniazid, the reference compound, exhibited a moderate clearance rate (Log CLtot = 0.782). Several derivatives showed clearance values close to this, suggesting that, despite structural modifications, the pyridine-4-carbohydrazide core structure contributes to a baseline level of metabolic stability. Derivatives with higher clearance rates (Log CLtot> 0.800), represented in Tuscan color, include INH05, INH06, INH10, INH11, INH14, and INH18. These compounds are cleared more rapidly from the body, indicating potentially shorter half-lives and a possible need for more frequent dosing. Structural features such as electron-withdrawing groups (e.g., nitro groups in INH10 and INH11) or bulky aromatic substituents (e.g., the naphthyl group in INH14) likely enhance the compounds' interaction with metabolic enzymes, leading to increased clearance. In contrast, compounds INH01, INH02, INH03, INH07, INH08, INH09, INH12, INH13, INH15, INH17, INH19, and isoniazid exhibit moderate clearance rates (Log CLtot ~ 0.700–0.800), indicated in green color. These derivatives typically feature less complex aromatic substitutions, which may reduce metabolic enzyme interactions, resulting in slower clearance rates. Compounds with moderate clearance rates are likely to remain in the body for a longer duration, which may enhance their ability to reach and maintain therapeutic levels at target sites. The lowest clearance rates (Log CLtot< 0> ConclusionIn drug discovery, understanding and predicting the physicochemical properties of compounds is essential to optimize their pharmacokinetic profiles. The pyridine-4-carbohydrazide derivatives examined in this study show a clear trend of improved physicochemical properties compared to the parent compound, isoniazid (INH). These enhancements are attributed to the structural modifications involving diverse aromatic substituents, resulting in derivatives with varied molecular weight, volume, hydrophobicity and hydrogen bonding capacity. Crucially, all examined compounds meet the criteria of Lipinski's Rule of Five and the Veber rule, suggesting they possess favorable drug-likeness characteristics, high permeability and biological availability. Despite their increased molecular complexity, these compounds maintain drug-likeness without violations, reinforcing their potential as drug candidates. The bioactivity scores of the derivatives demonstrate that structural modifications significantly influence predicted interactions with various biological targets. Unlike isoniazid, which exhibits limited bioactivity across a range of targets, derivatives with aromatic electron-donating groups showed improved bioactivity. On the other hand, the presence of electron-withdrawing groups tends to diminish the compounds' biological activity. This highlights the importance of careful structural modification to optimize bioactivity profiles, as the electronic properties of these groups directly impact target binding affinities. The PASS predictions for the pyridine-4-carbohydrazide derivatives provide key insights into the impact of structural modifications on their pharmacological potential. Compounds such as INH03, INH09, INH14 and INH19 show high predicted activity across various therapeutic categories, positioning them as strong candidates for further experimental investigation. These derivatives demonstrate potential in antibacterial, antiviral, antiprotozoal, anti-inflammatory and anticancer applications. The observed structural modifications create opportunities for the discovery of novel bioactive agents, offering promising avenues for the development of new therapeutic interventions across a range of medical fields. The pharmacokinetic profiles of the pyridine-4-carbohydrazide derivatives also highlight the influence of structural modifications on absorption, distribution, metabolism and excretion. Many of the derivatives show improved absorption and permeability profiles compared to isoniazid, particularly those with electron-withdrawing or hydrophobic substituents. However, some derivatives face challenges such as limited solubility or interactions with P-glycoprotein (P-gp), necessitating further optimization to overcome these absorption barriers. In terms of distribution, bulky and lipophilic derivatives exhibit greater tissue penetration and blood-brain barrier permeability, suggesting their potential utility in treating CNS-related conditions. However, these modifications may also increase the risk of CNS side effects, requiring a balanced approach to design. Metabolic profiling highlights the influence of specific substituents on interactions with cytochrome P450 enzymes, with some derivatives, such as INH14, exhibiting enhanced CYP450 inhibitory activity. This underscores the need to carefully consider structural modifications to minimize drug-drug interaction risks. Excretion rates also vary, with derivatives showing different clearance values. Those with higher clearance rates may require more frequent dosing, while others with lower clearance could risk accumulation. The absence of renal OCT2 interactions suggests hepatic clearance pathways, reducing the risk of renal toxicity. Overall, while these derivatives show promising pharmacological and pharmacokinetic profiles, further optimization and experimental validation will be essential to fully realize their therapeutic potential. Declaration of Competing InterestAll authors have made significant contributions to this manuscript, as follows: (a) conception, design, analysis, and interpretation of the data; (b) drafting the manuscript or critically revising it for significant intellectual content and (c) approving the final version to be published. Sofian S. Mohamed led the study’s design, investigation, data interpretation, and initial drafting.Salah M. Bensaber provided supervision and contributed to the study design.Nisreen H. Meiqaland Anton Hermann participated in reviewing and editing the manuscript. Abdul M. Gbaj contributed through supervision, validation and drafting of the manuscript. All authors have reviewed and approved the final version of the manuscript. The authors confirm that this manuscript has not been submitted to, nor is it under review at, any other journal or publication venue. AcknowledgmentsWe would like to thank the team of Judicial Expertise and Research Center, Tripoli, Libya for technical support throughout this project. We also thank Sir consultant Khaled Abuajila Diab (CEO of Judicial Expertise and Research Center, Tripoli, Libya) for his valuable support while performing this project. Ethical approvalAll the methods were performed under relevant institutional guidelines and regulations. Appendix A. Supplementary materialAppendix A. Supplementary material Supplementary data associated with this article can be found in the online at [XXXXXX]. Supplementary material Table 4A: Physicochemical Properties and Drug-Likeness Scores for the Predicted Compounds.
Abbreviations: Number of nonhydrogen atoms (N atoms); Molecular Weight (MW); Logarithm of partition Coefficient Between n-octanol and water LogP); Number of hydrogen bond acceptors (N-ON,O, and N atoms); Number of Hydrogen Bond Donors (N-OHNH,OH, and NH groups); Number of Rotatable Bonds (N-rotb),Topological Polar Surface area (TPSA); and Number of violations (N violations); Number of violations (N violations); and Molecular volume (Å3). Table 4B: Bioactivity score of compounds according to Molinspiratin cheminformatics.
Abbreviations: G protein-coupled receptor (GPCR) ligand, Ion channel modulator (ICM), Kinase inhibitor (KI), Nuclear receptor ligand (NRL), Protease inhibitor (PI), Enzyme inhibitor (EI). The gray color represents inactive and the green color represents moderate bioactivity under the scores reflect the predicted bioactivity, with more negative values indicating lower predicted activity for a given target. Table 5: PASS prediction Properties of the Predicted Compounds.
Abbreviations: Tuscan shading indicates the probability of activity values, and Gray represents the probability of inactivity values. Table 6A: Absorption Properties of the Predicted Compounds.
Abbreviations: Water solubility (logS, log mol/L), Human Intestinal Absorption (HIA, %), Human colon epithelial cancer cell line (Caco-2, Log Papp; log cm/s), Skin permeability (LogKp; cm/h), Permeability glycoprotein I, II (P-gp I, II). Gray shading indicates low values, green represents moderate values, and Tuscan signifies high values. Pink shading and "Yes" denote an effect on the target, while white shading and "No" indicate no effect on the target. Table 6B: Distribution Properties of the Predicted Compounds.
Abbreviations: Distribution Volume in Humans (VDSS, Log L/kg), Fraction Unbound (FU), Blood blood-brain barrier Permeability (LogBB), and Central Nervous System Permeability (LogPS). Gray shading indicates low values, green represents moderate values, and Tuscan signifies high values. Table 6C: Metabolism Properties of the Predicted Compounds.
Abbreviations: CYP (Cytochrome P450), Inhi (Inhibitor), Subs (Substrate). The pink color and "Yes" indicate an effect on the target, while the white color and "No" indicate no effect on the target. Table 6D: Excretion Properties of the Predicted Compounds.
Abbreviations:Total clearance (CLtot),Organic cation transporter 2 (OCT2). The gray color represents low, the green color represents moderate, and the Tuscan color represents high clearance in accordance with the value by Log ml/min/kg. References

Virginia E. Koenig

Delcio G Silva Junior

Ziemlé Clément Méda

Mina Sherif Soliman Georgy

Layla Shojaie

Sing-yung Wu

Orlando Villarreal

Katarzyna Byczkowska

Anthony Kodzo-Grey Venyo

Pedro Marques Gomes

Bernard Terkimbi Utoo

Prof Sherif W Mansour

Hao Jiang
Dr Shiming Tang

Raed Mualem

Andreas Filippaios

Dr Suramya Dhamija

Bruno Chauffert

Baheci Selen

Jesus Simal-Gandara

Douglas Miyazaki

Dr Griffith

Dr Tong Ming Liu

Husain Taha Radhi

S Munshi

Tania Munoz

George Varvatsoulias

Rui Tao

Khurram Arshad

Gomez Barriga Maria Dolores

Lin Shaw Chin

Maria Dolores Gomez Barriga

Dr Maria Dolores Gomez Barriga

Dr Maria Regina Penchyna Nieto
Dr Marcelo Flavio Gomes Jardim Filho

Zsuzsanna Bene

Dr Susan Weiner

Lin-Show Chin

Sonila Qirko

Luiz Sellmann

Zhao Jia

Thomas Urban

Cristina Berriozabal

Dr Tewodros Kassahun Tarekegn

Dr Shweta Tiwari

Dr Farooq Wandroo

Dr Anyuta Ivanova

Dr David Vinyes

Gertraud Teuchert-Noodt

Dr Elvira Farina

Dr Oleg Golyanovski | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||