
Case report | DOI: https://doi.org/10.31579/2578-8868/307
1M4 student Virginia Commonwealth University School of Medicine.2Department of Neurology, Virginia Commonwealth University Health System 11958 W. Broad Street, 4th floor, Henrico, VA 23233, USA.
*Corresponding Author: Xinli Du, MD PhD Department of Neurology, Virginia Commonwealth University Health System 11958 W. Broad Street, 4th floor, Henrico, VA 23233, USA.
Citation: Huang He Ding, Xinli Du, (2024), Rituximab in the Treatment of LRP4-Antibody-Positive Myasthenia Gravis: A Case Report, J. Neuroscience and Neurological Surgery, 15(2); DOI:10.31579/2578-8868/307
Copyright: © 2024, Xinli Du. This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
Received: 29 February 2024 | Accepted: 15 March 2024 | Published: 26 March 2024
Keywords: myasthenia gravis; lrp4 autoantibody; rituximab; efficacy
Antibodies against LRP4 were found in 2-4% of patients with myasthenia gravis (MG). LRP4-antibody-positive (LRP4+) MG presents with more severe disease with bulbar involvement. No standard treatment guideline has been established for this subtype. We identified a LRP4+MG patient with severe symptoms including bulbar dysfunction that were refractory to standard therapy. Rituximab was started due to treatment failure with standard therapy. At the time of initiation of rituximab, patient was on plasmapheresis every 1-2 weeks. Her work and quality of life were greatly compromised. Six weeks after initiating rituximab therapy, remarkable improvements in dyspnea, dysphagia, ptosis, and proximal limb muscle strength were reported without adverse events. Improvements in 3 standardized Myasthenia Gravis scoring systems were observed. The maintenance plasmapheresis was discontinued and pyridostigmine dose decreased by 50% because of sufficient symptom control. Stable improvement was achieved with rituximab maintenance therapy spaced 5 months apart. Our case demonstrates rapid onset of efficacy of rituximab in patient with refractory LRP4+MG. This response and the proposed mechanism of action of LPR4-Ab suggest that the efficacy of rituximab in LRP4+MG may parallel that in MuSK+MG. Further exploration of this treatment option is warranted.
Myasthenia gravis (MG) is an antibody-mediated autoimmune disorder affecting the neuromuscular junction (NMJ) function leading to fluctuating weakness of ocular, facial, bulbar, limb and respiratory muscles.[1] Approximately 80% of patients with generalized MG have antibodies targeting the acetylcholine receptors (AChR), and about 10% have antibodies against the muscle specific kinase (MuSK) protein.[2] Among the remaining seronegative MG patients, about 15% have antibodies recognizing lipoprotein-related protein 4 (LRP4) and/or agrin.[3] Although majority of MG patients respond to standard therapies including cholinesterase inhibitors, corticosteroids, steroid-sparing immunosuppressants such as mycophenolate mofetil and azathioprine, up to 15% patients still experience insufficient control of symptoms or intolerable side effects.[4,5] Identification of MG autoantibodies is a key step, as it not only helps with confirming diagnosis but also guides management of MG. For example, complement inhibitors are indicated only for AChR+MG based on the finding that complement activation and subsequent destruction of postsynaptic membrane by the membrane attack complex play a critical role in pathogenesis of AChR+MG;[6,7,8] thymectomy is effective in young AChR+MG but not in MuSK+MG as thymic hyperplasia rarely occurs in MuSK+MG;[10] rituximab is highly efficacious in MuSK+MG.[9]
LRP4 autoantibodies are believed to be pathogenic for MG by disrupting activation of MuSK.[11] LRP4+MG patients tend to report more severe disease with increased bulbar involvement similar to MuSK+MG patients. LRP4+MG patients are oftentimes managed with standard therapy due to lack of studies on effective treatment regimen.[3] We report a case of LRP4+MG patient with a refractory disease course who had excellent response to rituximab.
The patient presented in this case is a 54-year-old Caucasian female with relevant past medical history of depression, recurrent pulmonary infections (on chronic prophylactic antibiotics). She presented in her late 20s with muscle fatigue, ptosis, and dyspnea followed by dysphagia, at one point requiring parenteral feeding. Her initial work up included normal routine EMG, normal repetitive nerve stimulation studies, and negative AChR and MuSK autoantibodies. Her initial single fiber EMG (SFEMG) was nondiagnostic due to poor cooperation. She was empirically diagnosed with MG based on her clinical presentation and treatment response to pyridostigmine. Her MG symptoms worsened shortly after the diagnosis that led to multiple admissions for MG crisis and required chronic non-invasive ventilation at night and short periods during the day due to insufficiently controlled dyspnea and de-saturation. Prednisone offered some benefits but caused severe depression and avascular necrosis in the right hip. She received IVIG every 2-4 weeks for ~3 years and was switched to plasmapheresis (PLEX) due to frequent worsening of MG symptoms leading to numerous admissions for respiratory failure. Due to her history of recurrent pulmonary infection, her pulmonologist was resistant to chronic immunosuppression. She continued PLEX for 6 years; initial frequency every 3 weeks, increased to every 1-2 weeks due to insufficient control of bulbar symptoms. She continued to have frequent ED visits and admissions due to episodic worsening of MG symptoms. Repeat SFEMG of the extensor digitorum communis 2weeks following PLEX demonstrated increased jitter in 3/20 pairs with evidence of blocking. LRP4 antibody was subsequently checked and found to be positive for 2 times. Rituximab at 1000mg spaced by 14 days were initiated after consultation with her pulmonologist. The patient tolerated the infusion well. Six weeks after rituximab infusion, she noted significant improvement in dyspnea, dysphagia, ptosis, and proximal limb muscle strength. Plasmapheresis was tapered off and the daytime pyridostigmine dose was decreased by 50%. Evaluation at 11 weeks post initiation of rituximab infusion demonstrated 11-point improvement in the Myasthenia Gravis Activities of Daily Living profile (MG-ADL), 4-point improvement in Myasthenia Gravis Quality-of-Life 15 scale (MG-QoL15r), and 11-point improvement in Myasthenia Gravis Composite scale (MSC) (Fig 1). She no longer needed respiratory support during the day and reported greater tolerance of physical activities. She was exacerbation-free for 4.5 months, at which point rituximab therapy was repeated with similar clinical response. She had no pulmonary infections since initiating rituximab.
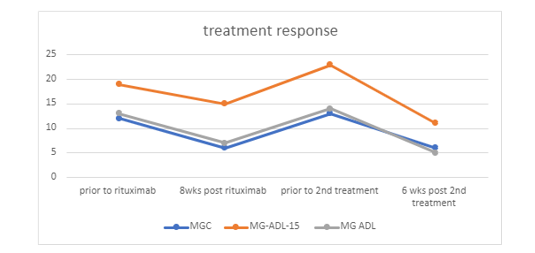
Figure 1: MGC, MG-ADL 15, MG ADL scores before and after Rituximab infusion.
The identification of antibodies is a key step in the workup of myasthenia gravis, as it not only confirms the diagnosis and can dictate the appropriate treatment. Common therapeutic options such as early referral for thymectomy should be considered for AChR+MG in patients younger than the age of 50, while newer therapeutics such as complement inhibitors and FcRn antagonists are indicated only for AChR+MG.[5,10] The International Consensus Guidance for Management of Myasthenia Gravis 2020 update recommends rituximab be considered as an early therapeutic option in patients with MuSK+MG who have an unsatisfactory response to initial immunotherapy, while in refractory AChR+MG, the efficacy of rituximab is uncertain and only be considered if patients fail or do not tolerate other immunosuppressant agents.[12]
LRP4+MG is a rare subtype of MG. It is believed that LRP4 interacts with agrin and MuSK to activate the kinase and initiates downstream signaling cascades for AChR clustering.[13] Recent work in mice has demonstrated that the presence of anti-LRP4/agrin antibodies in MG patients are pathogenic through disruption of agrin-dependent LRP4-MuSK interaction.[11]
No established treatment guidelines exist for LRP4+MG and there are few published reports of effective treatments. One multicenter case series study has demonstrated that patients with LRP4 antibodies experience more severe symptoms than those with double seronegative MG, and that LRP4+MG are generally managed with conventional immunosuppressant therapy.[3] The study also had 11% of its participants undergone treatment with rituximab, although the treatment response was not specified.[3] Our case demonstrates rapid onset of efficacy along with persistent action for a duration of 4.5 months in a patient with refractory LRP4+MG following rituximab infusion. Similar benefits were observed with maintenance infusion every 5 months.
Considering the pathophysiologic mechanism of LRP4 and its close interaction with the MuSK protein, the efficacy of selective B-cell depletion agents such as rituximab in LRP4+MG may be similar to that in MuSK+MG.[3,13] As a case report, extrapolation of this single data point is not feasible. Being a small subset of a rare disease, enrolment of enough patients to detect a significant therapeutic response is challenging. Many recent clinical trials of promising new MG therapies exclude sero-negative and LRP4+ patients, regardless of the potential efficacy. Publication of case reports of effective treatments in this population will help direct the development of a much-needed data-driven targeted algorithm for managing LRP4+MG.
Treatment for myasthenia gravis is rapidly evolving owing to the advance in identification of autoantibodies, understanding of the pathogenesis both at the neuromuscular junction and broadly the immune system, and the development of targeted therapeutics. Most of the new therapies are for AChR+MG. LRP4+MG represents a small portion but tends to have more severe clinical symptoms including bulbar dysfunction that can be refractory to standard therapies developed for AChR+MG. Guidelines and evidence-based practice specific for LRP4+MG are missing, as a result, obtaining approval for using non-standard therapy in patients with LRP4 antibody can be challenging. Our case report showed that rituximab had fast onset and persistent benefits in a LRP4+MG patient with severe and refractory disease. This opens the door for future research study of the efficacy of rituximab either by large case series or randomised control trial.
Both authors have no disclosure
It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,

Dear Maria Emerson, Editorial Coordinator of International Journal of Clinical Case Reports and Reviews, What distinguishes International Journal of Clinical Case Report and Review is not only the scientific rigor of its publications, but the intellectual climate in which research is evaluated. The submission process is refreshingly free of unnecessary formal barriers and bureaucratic rituals that often complicate academic publishing without adding real value. The peer-review system is demanding yet constructive, guided by genuine scientific dialogue rather than hierarchical or authoritarian attitudes. Reviewers act as collaborators in improving the manuscript, not as gatekeepers imposing arbitrary standards. This journal offers a rare balance: high methodological standards combined with a respectful, transparent, and supportive editorial approach. In an era where publishing can feel more burdensome than research itself, this platform restores the original purpose of peer review — to refine ideas, not to obstruct them Prof. Perlat Kapisyzi, FCCP PULMONOLOGIST AND THORACIC IMAGING.
