
case report | DOI: https://doi.org/10.31579/2692-9406/128
1Department of Neurosurgery, University of Florida, Gainesville.
*Corresponding Author: Brandon Lucke-Wold, Department of Neurosurgery, University of Florida, Gainesville.
Citation: Yusuf Mehkri, Brandon McDonald, Sai Sriram, Ramya Reddy, Brandon Lucke-Wold. et al (2022) Recent Treatment Strategies in Alzheimer's Disease and Chronic Traumatic Encephalopathy. Biomedical Research and Clinical Reviews, 7(3); DOI: 10.31579/2692-9406/128.
Copyright: © 2022 Brandon Lucke-Wold, This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 08 August 2022 | Accepted: 30 August 2022 | Published: 08 September 2022
Keywords: neurotrauma; alzheimer’s disease; chronic traumatic encephalopathy; pre-clinical models
Neurotrauma has been well linked to the progression of neurodegenerative disease. Much work has been done characterizing chronic traumatic encephalopathy, but less has been done regarding the contribution to Alzheimer’s Disease. This review focuses on AD and its association with neurotrauma. Emerging clinical trials are discussed as well as novel mechanisms. We then address how some of these mechanisms are shared with CTE and emerging pre-clinical studies. This paper is a user-friendly resource that summarizes the emerging findings and proposes further investigation into key areas of interest. It is intended to serve as a catalyst for both research teams and clinicians in the quest to improve effective treatment and diagnostic options.
Alzheimer’s Disease (AD) remains as the most prevalent cause of dementia worldwide and continues to be a significant health burden to society. Research in recent decades has significantly advanced our understanding of the pathophysiology of AD and its two neuropathological hallmarks: extracellular aggregations of insoluble forms of amyloid-β peptide (Aβ), known as Aβ or senile plaques, and intracellular neurofibrillary tangles (NFTs) predominantly comprised of hyperphosphorylated tau protein (p-tau) [1]. First hypothesized by Hardy and Higgins in 1992, the long-standing amyloid cascade hypothesis (ACH) postulates that senile plaques are the causative agent of AD and the neurodegenerative sequelae that follow [2]. While this is the most favorable explanation for familial AD, studies have yielded conflicting results as to whether Aβ plaques are the initiator or consequence of sporadic AD, but it is generally agreed that they play an unequivocal role in both forms of AD.
Discoveries in AD research further elucidated the potential mechanisms behind other neurodegenerative diseases; namely, chronic traumatic encephalopathy (CTE). CTE is a sporadic tauopathy associated with repetitive minor head trauma and is most frequently seen in athletes of high-impact sports, such as boxing or American football, and victims of domestic violence [3]. While usually clinically indistinguishable, CTE and AD are neuropathologically distinct. CTE is characterized by perivascular p-tau deposition in the sulcal regions of the cerebral cortex, found irregularly within surrounding neuronal cell and cell process NFTs [3]. By contrast, AD demonstrates a progressive but specific distribution pattern of Aβ plaques and p-tau NFTs that begins in the transentorhinal region early on and spreads to virtually all isocortical association areas by the terminal stages [4]. This distinction led to the recognition of CTE as a separate diagnosis from AD in dementia patients, although both conditions may only be confirmed by post-mortem examination [5,6].
Improvements in our understanding of the pathologic hallmarks of AD led to the development of multiple antibody drug therapies aimed at preventing or reducing Aβ plaque load in the brain, which have proven to be largely ineffective in altering the course of AD [7,8]. Like other early models for monoclonal antibody drugs, solanezumab targeted soluble forms of Aβ, which proved ineffective in two phase III trials [8]. A phase III clinical trial for gantenerumab, a human monoclonal antibody that instead binds to aggregated forms Aβ and removes plaques via Fc-receptor phagocytosis, did not demonstrate efficacy either and was terminated prematurely for futility [9]. Based on compiled statistical data from clinical trials, the most recent evidence suggests that a general reduction in Aβ plaques does not significantly improve cognition, suggesting that Aβ alone may not be the right target [10]. Or, as some newer articles theorize, we may be targeting the wrong hormetic form of Aβ [11].
Perhaps the most controversial drug is aducanumab, a monoclonal antibody targeting aggregated forms of both soluble and insoluble Aβ. The drug received accelerated FDA approval for the treatment of AD in 2021 [12]. despite conflicting results from its two phase III clinical trials and lack of established correlation between reduced plaque load and cognitive improvement [13]. The decision elicited a substantial outcry from the research community, and a case for the accelerated withdrawal of aducanumab was published by early 2022 [14]. Despite its controversy, the drug was found to have a downstream effect on p-tau levels in cerebrospinal fluid (CSF), which has major implications in the treatment of other neurodegenerative disorders aberrantly expressing p-tau, such as CTE [15,16]. These developments have led researchers to explore tau hyperphosphorylation and other biochemical mechanisms as potential targets for drug therapies in AD, CTE, and related neurodegenerative disorders.
Emerging clinical trials in Alzheimer’s Disease
Several emerging treatment targets for AD are currently under early investigation in clinical trials. Two central themes in AD pathology are amyloid aggregation and neuroinflammation, which together cause dysregulation of homeostasis and establish a neurotoxic environment. Here, we highlight some recent advances in the targeting of these two domains.
Targeting amyloid pathways
Amyloid-β oligomer (AβO) deposition is thought to promote neuronal death by disrupting mitochondrial and lysosomal membrane integrity,[17,18]. causing endoplasmic reticulum dysfunction, [18,19]. inducing a high intracellular calcium state, [20]. and promoting pro-inflammatory oxidative stress [21]. Targeting this amyloid pathway may, in turn, confer a neuroprotective benefit in AD patients. To this end, CT1812 is a small molecule sigma-2 receptor antagonist which is recently under investigation in a phase 2 trial for its role in synaptic AβO clearance [NCT03507790]. In preclinical studies, though treating cultured neurons with human AD-derived AβO led to vesicle trafficking deficits, these deficits were abolished when cultures were pretreated with CT1812.22 In addition, CT1812 improved synaptic density in neuronal mouse cultures by displacing AβO from receptors, facilitated AβO clearance into CSF in vivo mouse models, and improved cognitive performance in a mouse model of AD [22]. Promising data from a phase 1b trial of 19 patients receiving placebo or CT1812 for 28 days revealed improved AβO clearance into the CSF of patients receiving CT1812 [22]. However, higher-powered studies with longer treatment durations focusing on cognitive outcomes are necessary and are already underway.
Other variants of Aβ have recently come under interest as treatment targets. In particular, amyloid precursor protein (APP) can be cut by secretases and modified by glutaminyl cyclase to form pyroglutamylated forms of Aβ (pE-Aβ), a particularly cytotoxic variant of Aβ known to induce synaptic dysregulation [23,24]. Recently, the small molecule glutaminyl cyclase inhibitor PQ912 has been explored as a potential treatment of AD. One preclinical study of a transgenic AD mouse model treated with PQ912 displayed promising results, both decreasing pE-Aβ in vivo and improving cognitive performance [25]. Another recent phase 2 study of PQ912 of 120 patients randomized to PQ912 or placebo groups reported no significant adverse events (AE) for the PQ912 group, a significant reduction in glutaminyl cyclase activity, and a reduction in the glial activation marker YKL-40 [24]. Notably, though PQ912 was associated with improved performance on the one-back cognitive test, there were no differences between PQ912 and placebo groups in the mini-mental state examination (MMSE) scores [24]. A phase 2A study for use of PQ912 to assess cognitive function in early AD patients is underway [NCT03919162].
A novel proteopathy has been recently implicated in the progression of AD. Filamin A (FLNA) is a scaffolding protein thought to be altered in AD. Altered FLNA allows for Aβ and nAChR-mediated neurofibrillary tangle formation with activation of TLR4 and subsequent neuroinflammatory marker release [26]. PTI-125 is a small molecule which resets the altered variant of FLNA back to normal [26] and, in a transgenic AD mouse model, improved certain cognitive performance measures, reduced neuroinflammation, and decreased other biomarkers of AD progression [27]. In a recent phase 2a study consisting of 13 AD patients treated with 28 days of PTI-125, no drug related AE were observed [28]. Treatment with PTI-125 was appropriately associated with the reduced presence of altered conformation FLNA. Importantly, markers of AD progression including tau, neuroinflammatory cytokines such as IL-1B, IL-6, and TNFa, neurogranin, and neurofilament light chain were significantly reduced following 28 days of PTI-125 treatment in this study [28]. Though cognition was not evaluated in this study, a current phase 2b study seeks to add to previous work by randomizing a larger number of participants, assessing CSF markers of AD progression, and evaluating cognition [NCT04388254].
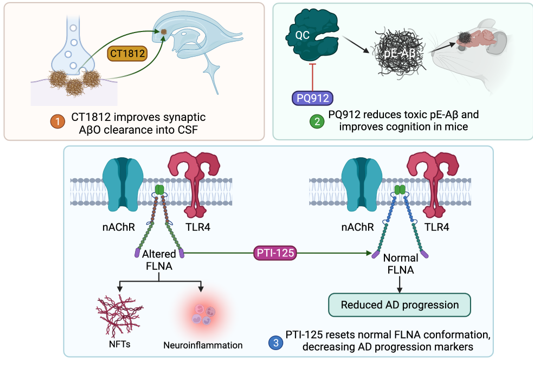
Figure 1: Molecular pathways of amyloid-targeting emerging therapeutics. Abbreviations: Aβ, amyloid β; pE-Aβ, pyroglutamylated forms of Aβ; QC, glutaminyl cyclase; TLR4, toll-like receptor-4; nAChR, nicotinic acetylcholine receptor; FLNA, filamin A; NFT, neurofibrillary tangle
Targeting neuroinflammatory pathways
Neuroinflammatory pathways have become an important drug target due to their prevalent role in the progression of AD [29]. One hallmark of AD is an elevated burden of fatty acid oxidation and synthesis. While fatty acids are crucial for proper brain and neuron function, high levels of fatty acid oxidation both modulates inflammatory processes and promotes neurodegenerative mechanisms [30,31]. CMS121 is a small molecule fatty acid synthase inhibitor which reduces oxidative damage by preventing excess lipid peroxidation [31]. Administration of CMS121 in neuronal and microglial cells as well as a transgenic AD mouse model led to decreased markers of lipid peroxidation and neuroinflammation. Spatial learning and memory tests of APPswe/ PS1ΔE9 transgenic mouse models treated with CMS121 also revealed improved cognitive function compared to wild type mice [31]. A phase 1 trial is currently underway to evaluate the long-term safety of this drug in healthy patients [NCT05318040].
AD has an impact on multiple pathways in the immune response. Therefore, treatments targeting the immune response from multiple angles may represent a more promising immunomodulatory approach. Recently, the antineoplastic TNFa inhibitor lenalidomide has come under interest for the treatment of AD due to its pleiotropic effects of modulating both innate and adaptive immune responses [32,33]. Notably, lenalidomide simultaneously lowers the expression of pro-inflammatory agents (TNFα, IL-6, and IL-8) and increases the expression of anti-inflammatory cytokines (IL-10) [33]. Preliminary results in vivo transgenic AD mouse models treated with lenalidomide showed significant decreases of key markers of AD progression including BACE1 and Aβ plaques as well as pro-inflammatory markers such as TNFa [33]. Despite this, special care should be exercised in further work considering some reports of lenalidomide-induced cognitive impairment [34]. A phase 2 trial is currently in progress to determine the efficacy of lenalidomide in reversing cognitive impairment in patients with mild to moderate AD [NCT04032626].
Despite the predominant view that neuroinflammation leads to progression of AD, recent work has investigated the pro-inflammatory cytokine granulocyte macrophage colony-stimulating factor (GM-CSF) as a potential treatment for AD. Interestingly, studies suggest that individuals with autoimmune diseases like rheumatoid arthritis (RA) have a lower risk of developing AD, [35] and GM-CSF has been shown to be elevated in patients with RA [36]. Sargramostim is an FDA approved recombinant human GM-CSF, and efforts have been made to determine the efficacy of this drug for the treatment of AD. Preclinical data of transgenic AD mouse models treated with sargramostim show improved cognitive function and increased activated microglia [37,38]. One phase 2 trial of 40 patients receiving either sargramostim or placebo for 3 weeks displayed its immunomodulatory effects, with upregulation of IL-6, IL-10, and TNFa, and downregulation of IL-8. Importantly, a statistically significant cognitive improvement was seen in the MMSE [37]. A safety analysis conducted in this study did not display any serious AE or amyloid changes on imaging [37]. Despite this, future clinical studies are underway to determine the long-term safety and tolerance of the drug on patients with mild to moderate AD [NCT04902703].
Alzheimer's: Preclinical studies and potential pathways warranting further investigation.
Alzheimer’s disease (AD) is biologically marked by β-amyloid plaques and Tau neurofibrillary tangles [39]. As such, efforts till now have largely targeted amyloid [40]. Other major targets, according to recent Phase 2 and 3 clinical trial therapies, focus on amyloid, synaptic plasticity/neuroprotection, Tau tangles, and neuroinflammation [41]. However, anti-amyloid strategies have been largely disappointing [40]. Several other pathways have come to light recently, warranting further investigation. These pathways include but are not limited to neuroinflammation, [42] oxidative stress, [43,44]. efects in mitochondrial dynamics and function, [45]. cholesterol and fatty acid metabolism, glucose energetic pathways impairments in the brain, [46,47] autophagy failure, [48] and others [49,50]. The following paragraphs will describe major pathways with broad highlights, in hopes of pointing out potential therapies in pre-clinical models of Alzheimer’s disease.
Inflammation, especially in the central nervous system, is an acute process that physiologically provides protection against infection, toxins, and injury [40,49]. However, when the balance between pro- and anti-inflammatory signals is disrupted, this can lead to chronic neuroinflammation, [51,53] which has been strongly linked to AD [54,55]. In a pro-inflammatory state, the increased levels of proinflammatory cytokines can cause mitochondrial stress via Aβ signaling as well as increase oxidative stress and subsequently, blood-brain barrier (BBB) permeability [56,57]. Moreover, a major pro-inflammatory cytokine, IL-18, leads to increased Cdk5 and GSK-3β, both of which are key players in Tau hyperphosphorylation [57]. While neuroinflammation may not trigger AD onset, it seems to play a significant role in exacerbating AD progression and/or symptoms via Aβ and Tau pathologies [58]. Recent pre-clinical studies have investigated curcumin, a polyphenol fund in turmeric, to be potently antioxidant and anti-inflammatory [59]. Further investigation is ongoing in terms of its role with regards to AD pathogenesis [59]. Another physiologic system, that if overactivated, can lead to AD symptoms is the complement system [60]. An overactive complement system influences Aβ, Tau, and APOE4, which can lead to AD progression [61,62]. Both pathways are propagated by non-neuronal supportive cells, which normally protect neurotransmission. However, in the setting of inflammatory cytokines (e.g.: IL-1β and TNF-α), the physiologic protection of neurotransmission is inhibited, leading to AD-related symptoms [52,63].
Another major focus of pre-clinical research is oxidative stress (OS), which has been linked to the prodromal phase of AD and not necessarily AD onset [64,65]. Increased OS, due to mitochondrial dysfunction, energy metabolism imbalances, or natural aging, can impair several key neuronal physiologic functions – namely, neuronal plasticity, cytoskeletal structure, and cellular communication [49,66-69]. Neurons, in specific, are quite sensitive to OS since the normal antioxidant is low [70]. Increased levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) leads to Tau phosphorylation and eventual destabilization of microtubules. This causes a major disruption in neuron polarity and intracellular trafficking, leading to decreased function of synapses [Munoz 32433996]. [66] In addition, Aβ is also tied to ROS and RNS and other mediators of OS (e.g.: NOX, TGF-β, NF-kβ, Nrf2). [64,71,72] Excess ROS causes oxidization of nucleic acids (which leads to lethal damage and promotes protein aggregation) as seen in post-mortem reports of AD patients [73]. OS is also tied to several other pathways and thus, is an area of further investigation.
One area that OS is linked to is glucose hypometabolism. High ROS levels reduces the concentration/availability of enzymes in the glycolytic cascade, leading to glucose hypometabolism [74-76]. In AD, this is confirmed via intracellular lesions in a subset of neurons with the highest metabolic requirements, that morphologically have elongated, thin axons and reduced or absent myelin sheaths [77]. Insulin signaling is another area of significant interest due to its association of AD, as Aβ oligomers bind insulin receptors, causing internalization of them [78-80]. The association of insulin signaling to AD is further supported by observing increased AD risk in populations affected by insulin-related comorbidities (e.g.: diabetes mellitus) [81]. Ultimately, altered insulin signaling can lead to neuroinflammation, which is tied to increased levels of Aβ and GSK3β, leading to Tau hyperphosphorylation, as previously mentioned [80,82,83]. Thus, insulin signaling is another area that warrants further investigation with relation to AD.
Furthermore, vascular dysfunction or cerebrovascular abnormalities are common comorbidities noted with AD, although the precise correlation with onset and/or progression of AD symptoms needs to be further elucidated [84]. The general principle is thought to stem from the altered homeostasis that arises from disrupted blood flow and/or pressure. This can lead to microfractures in the BBB also, which lends itself to neuroinflammation risk and eventual AD-related symptoms as previously mentioned [47,85-87]. In AD specifically, animal models link ROS to causing increased levels of advanced glycation end products (AGE) and their receptors (RAGE) in vasculature, which is linked to Aβ plaques. This is an active area of research, with drugs targeting AGE and/or RAGE being tested in clinical trials [41,88-91]. RAGE is also categorized as a pattern recognition receptor and is tied to innate immunity and inflammation. Lastly, increased levels of AGE and RAGE are tied to AD but are also found due to natural aging. This provides another clue as to why aging is a risk factor for AD [88,89,92].
Lastly, impaired autophagy has been linked to AD, based on animal models and AD patients [64]. APOE4 overexpression leading to Aβ accumulation within lysosomes causes neuronal death in the hippocampus, especially CA2/3 regions in animal models [93,94]. Furthermore, high levels of ROS inhibit autophagosome fusion with lysosomes [64]. These two pathways link impaired autophagy to AD. As such, current preclinical studies are further looking into restoring physiologic autophagosome clearance to curb the progression of cognitive symptoms [95]. All in all, AD pre-clinical research spans a wide array of topics, including altered neurotransmission and more, displaying the complex, multifactorial pathogenesis of AD, lending itself to multiple potential therapeutic targets in the future.
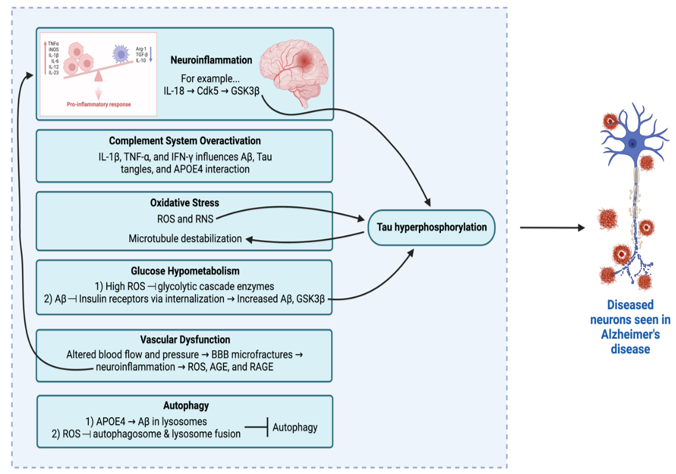
Figure 2: Summary of Select Pre-Clinical Alzheimer’s Disease Pathways
Chronic Traumatic Encephalopathy
Diagnostic and Management Considerations
Chronic Traumatic Encephalopathy (CTE) is a neurodegenerative condition characterized by an accumulation of hyper phosphorylated tau protein depositions throughout the brain.96 Microscopic and gross neuropathological changes originate from exposure to repetitive head impacts, leading to a progression of cognitive and neurobehavioral deficits [97]. Due to the unique neuropathological phenotype associated with CTE, conformational diagnostic strategies currently require post-mortem examination [98,99]. Overall, these diagnostic limitations present challenges for adequate management and therapeutic intervention. Thus, there is a critical need to improve diagnostic strategies through comprehensive psychiatric guidelines, imaging modalities, and biomarker analysis (Figure 3).
Neuropsychiatric Diagnosis and Symptomatic Management
Psychiatric evaluation of neurodegenerative diseases offers valuable preliminary diagnostic guidance. Previous studies have examined clinical features associated with CTE symptomatology to identify criteria correlated with cognitive and behavioral dysfunction [100,101]. Clinical guidelines were established in 2014 for diagnosis of Traumatic Encephalopathy Syndrome (TES), used as a proxy to describe the clinical symptoms of CTE [102]. Patients with TES present with progressive, cognitive deficits and/or neurobehavioral dysfunction associated with a prior history of repetitive head impacts [103]. Past exposure to repeated head trauma is of particular significance and the association between repeated mild TBI and neurodegenerative diseases is currently being investigated [NCT04124029]. Neurological deficits associated with TES can be classified into four symptomatic categories, including: cognitive, behavior, mood, and motor deficits [104]. Clinical assessments for these sequalae include evaluations for memory impairment and executive functioning, aggression and depression, and ataxia and dysarthria, respectively [100,105]. TES guidelines were validated for diagnostic efficacy in 2021, and researchers determined only cognitive symptoms were significantly associated with CTE [106]. However, these comprehensive criteria do provide useful methodology for excluding other neurological disorders and have been beneficial in establishing relevant management protocols. Current management strategies involve various forms of clinical intervention, including cognitive rehabilitation, behavioral therapy, as well as vestibular and ocular therapy [104]. Pharmacological agents have also been used for managing CTE, including stimulants, dopamine agonists, selective-serotonin reuptake inhibitors (SSRI’s [104]. These current management strategies have shown success for alleviating the cognitive and neurobehavioral deficits experienced clinically, however, none have shown efficacy for treating the disease. Thus, current limitations associated with TES guidelines must be overcome to ensure the development of effective therapeutic strategies.
Diagnostic Imaging Modalities
Several imaging modalities have been utilized to assess the diagnostic efficacy of examining neuropathological features associated with CTE. T1- and T2-weighted magnetic resonance imaging (MRI) are particularly useful in identifying gross structural changes associated with the disease [107]. Indeed, structural MRI indicated ventricular enlargement, cavum septum pellucidum and cortical atrophy were all significantly correlated with CTE [108]. Cortical atrophy was also shown to be correlated with AT8-immunostaining for tau pathology. Diagnostic efficacy for structural MRI is currently being investigated to determine how neurostructural changes are associated with chronic neurological outcome [NCT05235802]. In contrast, diffusion tensor imaging (DTI) may provide unique insight into identifying axonal integrity, and the dysfunction associated with repeated head impacts, specifically diffuse axonal injury (DAI) [109]. Using fractional anisotropy as a metric for evaluating axonal integrity, previous DTI studies showed that increased axonal disruption is associated with regions of hyperphosphorylated tau deposits [110]. Previous studies have also used imaging modalities to identify changes in metabolic profiles. Results from magnetic resonance spectroscopy (MRS) concluded metabolic changes in the anterior cingulate gyrus; including glutamate, glutathione, and myo-inositol, were all significantly correlated with behavior and mood domains [111]. Positron emission tomography (PET) has also been studied for confirming CTE diagnosis due to the molecular specificity associated with ligand binding. Former professional National Football League players were shown to have increased uptake in Flortaucipir in bilateral superior frontal, bilateral medial temporal and left parietal regions when compared against controls [100]. However, uptake was not significantly correlated with neuropsychiatric examinations. Other imaging molecules, including AV1451 and T807 have been validated for diagnosing CTE, but with variable success [112,113]. Currently, PET analysis using AV1451 is being investigation through the DIAGNOSE-CTE Research project, to be completed in 2023 [NCT02798185] [114].
Pathophysiological and Genetic Biomarkers
Fluid biomarkers, through collection of saliva, blood, and cerebrospinal fluid (CSF), have also been examined in patients suspected of CTE to assess diagnostic efficacy. Biomarkers associated with axonal injury, including tau, and neurofilament proteins light and heavy (NfL and NfH) have each been widely studied and associated with CTE [115]. Additionally, NfL has shown to be correlated with fractional anisotropy measurements from DTI studies, providing further rationale for usage in CTE diagnostics [116]. Other biomarkers including glial acidic fibrillary protein (GFAP) and s100b have also shown promise for validating pathophysiological features associated with CTE, specifically astrogliosis [117]. However, phosphorylated tau and amyloid beta remain primary diagnostic markers for discerning between CTE and AD [6]. Validation of fluid biomarkers is still an area of active investigation and current clinical trials are determining the proteomic and transcriptomic profiles associated with CTE [NCT04928534]. Additionally, genetic markers, including APOEε4 and TMEM106B may also be useful for diagnosing CTE, specifically for establishing links between genetic predispositions and CTE [118,119]. However, pathophysiologic, and genetic biomarkers alone do not possess adequate power for definitively diagnosing CTE. Thus, there remains a critical need to optimize and validate effective diagnostic and therapeutic protocols through pre-clinical studies.

Figure 3: Current diagnostic and management considerations for CTE. Cognitive and behavioral dysregulation associated with CTE was used to establish neuropsychiatric criteria for diagnosing TES, and symptomatic management for TES includes clinical and pharmacological intervention. Additionally, previous studies have validated the diagnostic efficacy for both imaging modalities and biomarkers and these strategies are currently under investigation in active clinical trials.
Preclinical Models
The development of preclinical animal models of CTE is challenging but necessary for the further investigation between the pathophysiological link between repetitive head trauma and chronic neurodegenerative tauopathy. While there are many different preclinical models of TBI, only a small subset has utilized repetitive and often mild injury to characterize chronic timepoints post-injury where the development of tauopathy is predicted to occur in CTE. Preclinical models of repetitive mild TBI (rmTBI) to investigate CTE are also a relatively recent development, with most experimental models being created in the past fifteen years. The experimental models that have investigated tau pathology following rmTBI have mainly been conducted in rodents utilizing diverse brain injury induction methods with or without genetic modifications in the tau gene, transgene expression of human tau (hTau), or overexpression of tau. Brain injury induction methods in rodent CTE models include both diffuse injuries, such as the fluid percussion injury (FPI), rotational injury to include the closed-head impact model of rotational acceleration (CHIMERA), and exposure to blast, and more focal brain injuries such as closed head injury (CHI), controlled cortical impact (CCI), or weight-drop models.
Diverse findings regarding rmTBI and tau pathology have been reported in preclinical models. Previous literature reviews have extensively covered preclinical CTE model findings from the years 2019 and prior [120-124]. Here, we have summarized here additional findings in CTE preclinical rodent models from 2020 to the present in Table 1. It is of interest to note that there is still conflicting evidence regarding presence and pathological severity of tauopathy following rmTBI among preclinical studies, with some studies identifying neuronal tau accumulation and others finding no differences in tau pathology. These differences are likely due to comparisons between models of different TBI injury paradigms and differences in TBI severity, timeline of repetitive injuries, and selected endpoints.
In modeling rmTBI with the goal of understanding the etiological link to CTE, there should be enhanced care taken in the field regarding consistency of parameters between models and a more stringent focus on translational capabilities of specific models. For example, rodent modeling of repeated rotational TBI may not be optimal in recapitulating the same forces and outcomes as human rotational brain injury due to the size of the rodent brain and lack of gyri and sulci [126,127], which are important not only in altering the way physical forces from the injury are distributed across the brain, but sulci and associated gray-white matter ratios are also highly associated with tauopathy in human patients [127,128]. Another important point in preclinical modeling of CTE is translatability in choice of brain injury induction method. Recent evidence has demonstrated that the risk of developing CTE is more associated with impact TBI, such as concussion events experienced in sports, versus blast TBI as experienced due to occupational exposure within the military [129]. Therefore, it may be critical to choose brain injury induction methods more associated with CTE like focal brain injury. Translation of preclinical research findings to better understanding of human CTE may also mean expanding the typical CTE model to new species whose brains are larger, gyrencephalic, and/or protected by a skull whose anatomy is more similar to humans [130,131]. Additional parameters to carefully consider in modeling CTE is the spacing of rTBI injuries, the age of the rodent during rmTBI, and the endpoint(s) at which pathology is assessed [102,122,132,133]. To better replicate the human timeline of rTBI, injuries should be spaced over a long period of time during adolescence to young adulthood, and chronic endpoints should be used for evaluation of CTE pathology. Currently, most models of rmTBI and CTE utilize short spacing of injuries and endpoints in rodents are typically chosen at 1-1.5 years of age, which only equates to mid-life in humans [134]. Overall, while preclinical studies of CTE represent one of the most favorable methods by which to investigate the pathophysiological link between rmTBI and CTE, there are key avenues to be addressed to enhance the translational capacity of preclinical models and enhance the investigation of causative mechanisms of CTE.
| Study | Animal | rTBI model | Injury Paradigm | Age | Tau Detection | Endpoint(s) | Findings |
| Tadepalli et al. 2020 [135] | Wild-type (WT) male rats | Mild weight drop (mWD) | Single mild (mTBI), re- petitive mild (rmTBI – 5 hits, 24 h apart), rapid repetitive mild (rapTBI – 5 hits, 5 min apart) or a single severe (sTBI) TBI | 8-10 months | Enzyme-linked immunosorbent assay (ELISA) | 8 weeks post-injury (PI) | No differences in phosphorylated tau (pTau); rmTBI group decreased performance in Novel Object Recognition (NOR) test |
| Cheng et al. 2020 [136] | Tau-overexpressing male mice (Tau58.4) | rmTBI via closed head impact (CHI) | 42 impact (6 impacts/day for 7 days) | 3-4 months | Immunohistochemistry (IHC) | 1 month PI | No differences in pTau, oligomeric Tau; rmTBI Tau-overexpressing mice had enhanced gliosis and peripheral immune cell infiltration |
| Bachstetter et al. 2020 [137] | WT or rTg4510 tauopathy male mice | CHI | Single or 2nd CHI exposured 1 day or 7 days PI | 4 months (WT), 2 months (rTg4510) | ELISA, Western Blot (WB) | 7 days PI (WT), 13 days PI (rTg4510) | Increased pTau and total Tau in 2 hit WT CHI mice; increased pS396/S404 positive tau in 1 and 2 hit rTg4510 CHI mice, no difference in total tau |
| Niziolek et al. 2020 [138] | Male mice with or without either i. pharmacological depletion of acid sphingomyelinase (Asm) or ii. genetic ablation of Asm (Asm-/-) | Moderate WD | Single | 8-10 weeks | IHC | 1 day or 30 days PI | TBI increased hippocampal pTau at 30 days PI and this accumulation is partially prevented by Asm inhibition |
| Tang et al. 2020 [139] | WT or transgenic Tau male mice (P301S) with or without Fyn kinase inhibition | CHI with or without chronic variable stress (CVS) | 14 hit (1 per day) over alternating hemisphere | 2 months | IHC | 5.5 months PI | Injury increased pTau but not total tau; Fyn inhibition rescued pTau levels to Sham |
| Angoa-Perez et al. 2020 [140] | WT male mice | WD | Exposed to a total of 20 head impacts (1 per day for 5 days [Monday-Friday with weekends off]) | 8 weeks | IHC | 0, 45, or 90 days PI | Increased pTau at 90 days PI |
| Bugay et al. 2020 [141] | WT male mice | Blast | 3 exposures, 1 per day over 3 consecutive days | 10 weeks | WB | 7 days PI | Increased tau and pTau in rmTBI group |
| Ojo et al. 2020 & Eisenbaum et al. [142,143]. | hTau mice | CHI | 2 injuries/week for 3 months | 3 months | Ex vivo tau uptake by cerebrovascular cells | 3, 6, or 12 months PI | Decreased tau uptake in rmTBI mice |
| Garcia et al. 2021144 | Male rats | Blast | 3 exposures, 1/day for 3 consecutive days | 10 weeks | WB | 10 days PI | Elevated pTau in blast animals and correlating neurobehavioral deficits |
| Dickstein et al. 2021 [112] | Male rats | Blast | 3 exposures, 1/day for 3 consecutive days | 10 weeks | WB, IHC | 6 weeks or 10 months PI | Elevated pTau at both endpoints in certain brain regions; no changes to total tau |
| Morin et al. 2021 [145] | hTau mice | CHI | Either i. 5 hit rmTBI (5 injuries over 9 days with a 48-hr interval) or ii. chronic rmTBI (24 injuries, 2 per week, with a 3-4 day interval) with or without 3 month delayed anatabine treatment for 3 months | 12-14 weeks | IHC | 6 months PI | Increased pTau in both rmTBI models; anatabine decreased pTau to sham levels |
| Xu et al. 2021 [146] | WT male mice | Closed skull controlled cortical impact (CCI) | 5 impacts, 1/day for 5 consecutive days | 8-10 weeks | IHC | 1, 4, and 10 weeks PI | Increased pTau at 4 and 10 weeks PI in rmTBI mice |
| Hiskens et al. 2021 [147] | WT male mice | WD | i) a single impact (1-IMP); ii) five total impacts (5-IMP); iii) 15 total impacts (15-IMP) | - | ELISA | 2 days or 3 months PI | No differences in pTau or tau pathology |
| Kahriman et al. 2021 [148] | WT male mice | CHI | 5 hits, 1/day for 5 consecutive days | 8-12 weeks | IHC | 1, 4, or 24 weeks PI | Increased pTau-immunoreactive astrocytes and neurons and cortical pTau at 4 weeks and 24 weeks PI |
| Kahriman et al. 2022 [149] | WT male mice | WD
| 5 hits, 1/day for 5 consecutive days | 9-12 weeks | IHC | 28 days PI | Increased tau pathology in rTBI mice |
| Wu et al. 2022 [150] | WT and Interleukin 1 Receptor 1 kockout (IL1R1-/-) mice | CHI | 3 hit, 1 per consecutive day over alternating hemisphere beginning with right | 38 days | WB, IHC | ~13 months PI | Increased tau hyperphosphorylation and aggregation in male neurons but not female neurons; no differences in total tau |
| Yoon et al. 2022 [151] | WT male mice | WD | 5 hits, 1 administered every 3 days | - | IHC | 3 days PI | Increased pTau in the olfactory bulb |
| Juan et al. 2022 [152] | WT mice | CHI | Single or 5 hits administered every 48 hours | 12 weeks | WB, IHC | 1 month PI | No tau pathology differences, but tau regulatory proteins significantly altered in rmTBI mice |
| Morin et al. 2022 [153] | hTau mice | CHI | 5 consecutive mTBI over 9 days (48 h interval between the injuries) | 3 months | Mass spectrometry of isolated cortical proteins and phosphoproteins | 3 or 24 weeks PI | Increased tau associated protein in rmTBI group along with alterations to phosphoproteins; same model previously demonstrated increased tau pathology up to 2 years PI |
Table 1: Literature on CTE preclinical rodent models. Search terms for table: (chronic traumatic encephalopathy) AND model* OR (repetitive traumatic brain injury) AND tau on Pubmed.gov; refined by years 2020-2022 with focus on tau pathological assessment
Alzheimer’s disease and CTE both have strong connections with neurotrauma. Emerging data has looked at preceding inflammatory cascades that contribute to disease progression. In this review, we highlighted the novel clinical trials, innovative pre-clinical studies, and shared mechanisms between the disease states. This review offers a concise resource for clinicians and research personnel.
Declarations of Interest: None
Funding: This research did not receive any grant from funding agencies in the public, commercial, or not-for-profit sectors.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
