AUCTORES
Globalize your Research

Research Article | DOI: https://doi.org/10.31579/2639-4162/244
Retired Urologist and Clinician, Reviewer of Articles for Journals, Medical Examiner Member of Royal College of Pathologists, London. United Kingdom.
*Corresponding Author: Anthony Kodzo-Grey Venyo, Retired Urologist and Clinician, Reviewer of Articles for Journals, Medical Examiner Member of Royal College of Pathologists, London. United Kingdom
Citation: Anthony Kodzo-Grey Venyo, (2025), Embolization of Urinary Bladder for The Treatment of Intractable Unresolving Visible Haematuria an Update, J. General Medicine and Clinical Practice, 8(1); DOI:10.31579/2639-4162/244
Copyright: © 2024, Anthony Kodzo-Grey Venyo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 23 November 2024 | Accepted: 09 January 2025 | Published: 16 January 2025
Keywords: ochoa syndrome; hydronephrosis; chronic kidney disease; urofacial syndrome; urinary incontinence
It has been iterated that the Urofacial or Ochoa Syndrome (UFS or UFOS) is typified by an inverted facial expression (those affected seem crying while smiling) that is associated with lower urinary tract dysfunction without evident obstructive or neurological cause. It is stated to be associated with autosomal recessive inheritance mutations within the HPSE2 gene, that is located at 10q23-q24, and the LRGI2 gene, located in 1p13.2; nevertheless, in up to 16% of patients, no associated mutations had been identified. Recent evidence had indicated that these genes are critical to an adequate neurological development to the lower urinary tract and that the origin of the disease appeared to be related to peripheral neuropathy. There is clinical variability among patients who are afflicted by UFS and not all patients with UFS do manifest with the classic two components, and it has even been genetically confirmed in patients with a prior diagnosis of Hinman Syndrome or other urinary bladder dysfunctions. Also, the presence of nocturnal lagophthalmos in these patients had been recently reported. Early recognition and timely diagnosis of UFS are critical for the prevention of complications such as urinary tract infections or chronic kidney disease. Next, to support readers throughout the world about this difficult and enigmatic condition to elucidate, an update of the literature on UFS has been extensively provided in the article.
Ochoa Syndrome which is also referred to as Urofacial syndrome (USF) is iterated to have been first described in many unrelated Colombian families (Elejalde, 1979 [1]; Ochoa et al. 1987 [2] The Urofacial Syndrome or Ochoa syndrome is a very uncommon clinical condition, which generally because of its rarity would tend to be unknown by a large number of clinicians and medical care workers as well as patients. Ochoa Syndrome is characterised by, an inverted facial expression, emanating from abnormal contraction of facial and ocular muscles, especially when smiling, in addition to the presence of urinary tract system abnormalities. Individuals who have Ochoa Syndrome, usually tend to present with urinary bladder voiding dysfunction due to a lack of coordination between detrusor muscle contraction and urethral sphincter relaxation. This usually tends to be ensued by the development of enuresis, urinary tract infection and hydronephrosis, leading to renal damage and eventually kidney failure. The facial abnormality is a characteristic expression such that, when these patients smile, their facial musculature inverts and they do appear to be crying. This grimacing is highly diagnostic of the UFS. In addition to facial and urinary tract organ abnormalities, constipation had been reported in about two thirds of the patients (Ochoa et al. 1987). [2] The occurrence of Ochoa Syndrome in multiple siblings with normal parents, the equal sex distribution, and the increased parental consanguinity had indicated autosomal recessive inheritance for this condition. Utilising a combination of homozygosity-mapping and DNA pooling-strategies, Wang, et al. (1997) [3] were able to place the Ochoa Syndrome gene within a 1-cM interval on chromosome 10q23-q24. Mutation analysis had subsequently excluded the glutamate oxaloacetate transaminase gene (GOT1), located within this region, as a candidate gene for Ochoa Syndrome (Wang, et al. 1999). [4] Ochoa Syndrome is a rare condition of which the genetic background had recently been discovered. The polypeptide heparanase-2 which is responsible in facial expression and urinary voiding control. LRIG2 encodes polypeptides which do have role in neural signalling. [5] [6] Ochoa Syndrome is stated to be associated with autosomal recessive inheritance mutations within the HPSE2 gene, which is located at 10q23-q24, and the LRGI2 gene, located in 1p13.2; however, in up to 16% of patients, no associated mutations had been found. [7] Recent evidence had suggested that that these genes are critical to an adequate neurological development to the lower urinary tract and that the origin of the disease does seem to be due to peripheral neuropathy. There is clinical variability among patients who have Ochoa Syndrome and not all individuals do present with the classic two components of urinary and bowel problems in association incongruity of their facial expressions, and Ochoa Syndrome had even been genetically confirmed in patients who have had a prior diagnosis of Hinman Syndrome or other urinary bladder dysfunctions. Also, the presence of nocturnal lagophthalmos in patients who have Ochoa Syndrome had recently been reported. Early recognition and timely diagnosis are critical for the prevention of complications that are associated with urinary tract infections or chronic kidney disease as well as eye diseases. Considering that just about one hundred and fifty cases of Ochoa Syndrome had so far been reported in the literature, it would be envisaged that majority of clinicians all over the world would tend not to be familiar with the presentations, diagnosis and treatment associated with Ochoa Syndrome. The ensuing article on Ochoa Syndrome is divided into two parts: (A) Overview of the general aspects of Ochoa Syndrome and (B) Miscellaneous Narrations and Discussions from Some Case Reports, Case Series and Studies Related to Some Cases of Ochoa Syndrome.
To review and update the literature on Ochoa Syndrome
Internet data bases were searched including Google; Google Scholar; Yahoo; and PUBMED. The search words that were used included: Ochoa syndrome, Uro-facial Syndrome; Uro facial Ochoa syndrome; Hydronephrosis with peculiar facial expression; Hydronephrosis-inverted smile; Inverted smile and occult neuropathic bladder; Inverted smile-neurogenic bladder; Partial facial palsy with urinary abnormalities; and UFS. Fifty-seven (57) references were identified which were used to write the article which has been divided into two parts: (A) Overview which has discussed general overview aspects of Ochoa syndrome and (B) Miscellaneous narrations and discussions from some case reports, case series, and studies related to Ochoa Syndrome.
Results
[A] OVERVIEW
General Statements
• Ochoa syndrome is a terminology that is utilised for a disorder which is characterized by urinary problems as well as unusual facial expressions. [8]
• The urinary tract problems tend to be associated with Ochoa syndrome.
• It has been stated that Ochoa syndrome typically tends to become apparent during early childhood or adolescence. [8]
• Individuals who have Ochoa syndrome may have difficulty with regard to the control of the flow of their urine including urinary incontinence, which could result in bedwetting. [8]
• People who have Ochoa syndrome may not be able to completely empty their urinary bladder, which often tends to be followed by the development of vesicoureteral reflux as well as hydroureter and hydronephrosis. [8]
• Vesical-ureteric reflux and hydronephrosis could be followed by the development of recurrent urinary tract infections and pyelonephritis which might lead to the development of kidney failure. [8]
• Individuals who are affected by Ochoa syndrome also do exhibit a characteristic frown-like facial grimace when they try to smile or laugh, and this is often described as inversion of facial expression. It has been documented that while this feature might manifest earlier than the presentation of urinary tract symptoms, perhaps as early as an infant begins to smile, it is often not brought to the attention of medical practitioners. [8]
• About two-thirds of individuals who are affected by Ochoa syndrome also do experience problems with their bowel function including: constipation, loss of bowel control, or muscle spasms of the anus. [8]
• It has been iterated that when individuals who have been affected by Ochoa syndrome attempt to smile, the patients do appear to be crying or grimacing. [9]
• It has been stated that Ochoa syndrome, was first described by a Colombian physician called Bernardo Ochoa in the early 1960s. [9]
• It has been iterated that the inverted facial expression that is manifested by children with Ochoa syndrome does enable early detection of the syndrome, which is vital for the establishment of a better prognosis as urinary related problems associated with this disease can cause harm if they are left untreated. [9]
• Incontinence is stated to be another easily detectable manifestation of Ochoa syndrome which is due to detrusor-sphincter dyssynergia. [9].
• Ochoa syndrome is usually an autosomal recessive congenital disorder [9] [10], as well as Ochoa Syndrome might also be associated with HPSE2 [9] [11]
Terminology
Ochoa syndrome is also referred to by other names including the ensuing:
• Uro-facial Syndrome. [9]
• Urofacial Ochoa syndrome. [8]
• Hydronephrosis with peculiar facial expression [8] [9]
• Hydronephrosis-inverted smile [9]
• Inverted smile and occult neuropathic bladder [8] [9]
• Inverted smile-neurogenic bladder [8]
• Partial facial palsy with urinary abnormalities [8] [9]
• UFS [8] [9]
Frequency [9]
• It has been iterated that Ochoa Syndrome is an uncommon condition and that 150 cases of Ochoa syndrome had been reported in the medical literature. [8] [9]
Causes
The ensuing summations had been made related to the cause of Ochoa Syndrome: [8] [9]
• Ochoa syndrome could be caused by mutations in the HPSE2 gene.
• HPPSE2 gene does provide instructions for making a protein called heparanase 2.
• The function of this protein has not been well understood.
• Mutations within the HPSE2 gene that cause Ochoa syndrome, lead to changes within the heparanase 2 protein which likely prevent it from functioning.
• The connection between HPSE2 gene mutations and the features of Ochoa syndrome had not been clarified.
• In view of the fact that the areas of the brain which control facial expression and micturition are in close proximity, some researchers had postulated that the genetic changes might lead to an abnormality within this brain region which may account for the symptoms of Ochoa syndrome.
• Other researchers had conjectured that a defective heparanase 2-protein may lead to problems with the development of the urinary tract or with muscle function in the face and bladder.
• It has been iterated that some people who are affected by Ochoa syndrome do not have mutations in the HPSE2 gene. In these individuals, the cause of the disorder is not known.
Manifestations
The signs and symptoms of Ochoa Syndrome had been summated as follows: [9]
• Infants who are affected by Ochoa syndrome do exhibit an inverted smile; and they appear to be crying when they are actually smiling, in addition with symptoms of uropathy.
• Individuals and infants who are affected by Ochoa Syndrome might be afflicted by hydronephrosis.
• Symptoms of Ochoa syndrome could begin at very young ages.
• Many individuals who have Ochoa Syndrome would die within their teenage years to early 20s because of their kidney failure (uropathy) if the condition is not diagnosed and treated.
• Children who are afflicted by Ochoa Syndrome do have abnormal facial development which cause an inverted smile, but their nerve connections are however, normal.
• When a child who is affected by Ochoa Syndrome is attempting to smile, the child would tend to appear to be crying.
• In individuals who are affected by Ochoa Syndrome, urinary problems do arise as a result of a neurogenic urinary bladder.
• Majority of patients who are affected by Ochoa syndrome who are older than the age of toilet training, do present with enuresis, recurrent urinary tract infections, hydronephrosis, as well as a spectrum of radiology imaging abnormalities that are typical of obstructive or neurogenic urinary bladders. Some of the radiology imaging abnormalities do include the ensuing features:
Trabeculated urinary bladder
Vesicoureteral reflux.
External sphincter spasm.
Pyelonephritis,
Hyperreflexic urinary bladder, non-inhibited detrusor contraction, etc.
Urinary abnormalities might emanate in the development of renal deterioration and failure. This could be prevented by taking proper measures to restore normal voiding and by taking antibiotics to prevent infections.
In some cases, the afflicted patients do become hypertensive and they do progress to end-stage renal disease, while others become uremic. In addition, majority of patients are stated to suffer from constipation. [3]
It has been iterated that early detection of Ochoa syndrome is possible through the peculiar faces that children manifest with
Cause of Ochoa Syndrome
The ensuing summations had been regarding the aetiology of Ochoa Syndrome: [9]
• It has been stated that Uro-facial syndrome does occur as a result of either disruption or mutation of a gene on chromosome 10q23q24. [9] [12]
• The gene is stated to be located upon a 1 centimorgan interval between D10S1433 and D10S603 [3] [9].
• Alteration of this gene leads to alteration of facial and urinary developmental fields. This gene is understood to be the HPSE2 gene.
• It has been iterated that the HPSE2 gene is expressed within both the central nervous system as well as the urinary bladder.
• Heparanase 2 is a protein coded by exons 8 and 9 on the HPSE2 gene. This protein is understood to be altered in the case of this syndrome [11]
• Studies undertaken on mice had suggested that HPSE2 has no enzymatic activity. [13]
• Mutations within the HPSE2 gene on chromosome 10q23q24 had been found to cause Ochoa Syndrome. This means the defective gene that is responsible for the disorder is located upon an autosome (chromosome 10 is an autosome), and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder are stated to both carry one copy of the defective gene, but usually they do not experience any signs or symptoms of the disorder. [9]
• The relationship between a defective HPSE2 gene and Ochoa syndrome is stated not to be clear [9]. There is a postulate that the genetic changes may lead to an abnormality within the brain region, evidence for this postulation is that the areas of the brain that control facial expression and micturition are in close proximity of each other. Other hypotheses had been postulated that the defective heparanase 2-protein might lead to problems that are associated with the development of the urinary tract or with muscle function in the face and the urinary bladder. [8] [9].
Treatment
The treatment of Ochoa syndrome had been summarized as follows: [9]
• The treatment of Ochoa syndrome does tend to vary based upon the condition and extent of the uropathy.
• To empty the urinary bladder regularly and clean intermittent urethral catheterization (CIC) could be undertaken.
• If the urodynamic study does demonstrate non-inhibited detrusor contractions, an anticholinergic medication should be provided in addition.
• In order to prevent the development of recurrent infections, especially in the case of vesicoureteral reflux, the treatment could also include utilisation of prophylactic antibiotics.
Epidemiology
• It has been documented that Urofacial (Ochoa) syndrome had received the Ochoa name because of the first person to describe it in 1987, called Bernardo Ochoa. [3]
[B] Miscellaneous Narrations and Discussions from Some Case Reports, Case Series, And Studies Related to Ochoa Syndrome
Ochoa et al. [2] had reported that between 1965 and 1986 they had seen 36 children who had enuresis and urinary tract infection in association with “inversion” of facial expression when they were laughing. Ochoa et al. [2] also reported that urological work-up of these patients had found typifying findings of mild neuropathic bladder in all cases, with severe urinary tract damage in most of them. Ochoa et al. [2] recommended that the clear association of distortion in facial expression and neuropathic urinary bladder with resultant damage to the genitourinary tract should prompt urological evaluation of individuals who manifest with “inversion” of facial expression. Ochoa et al. [2] had also reported that about two thirds of the patients also had moderate to severe constipation. Ochoa et al. [2] suggested the term urofacial syndrome to be used for this disorder. Ochoa et al. [2] in addition documented that the occurrence of the disorder in multiple siblings, normal parents, increased parental consanguinity, and equal sex ratio indicate autosomal recessive inheritance.
In 2004, Ochoa et al. [14] had reported that during the preceding 40 years, over 100 patients had been reported with dysfunctional lower urinary tract symptoms which were associated with a peculiar distortion of facial expression. This most unusual disorder was initially considered to be a local observation. Nevertheless, time had proven otherwise, since patients with this syndrome, had subsequently been reported from various countries throughout the world. This association of lower urinary tract and bowel dysfunction with an abnormal facial expression was named the urofacial (Ochoa) syndrome. Genetic studies had demonstrated that this condition is inherited as an autosomal recessive trait, and a potential gene had been mapped to chromosome 10q23-q24. There was also enough evidence to indicate that patients who are affected by this syndrome as well as those with sub-clinical neurological urinary bladder, occult neuropathic bladder, non-neurogenic or neurogenic bladder or Hinman syndrome, dysfunctional voiding, or dysfunctional elimination, may be affected by the same congenital disorder of neurological origin.
Osorio et al. [7] iterated that the Uro-facial or Ochoa Syndrome (UFS or UFOS) is typified by an inverted facial [removed]in that those who are afflicted by Ochoa syndrome, seem to be crying while smiling) associated with lower urinary tract dysfunction without evidence of obstructive or neurological cause. Osorio et al. [7] iterated that Ochoa syndrome is associated with autosomal recessive inheritance mutations in the HPSE2 gene, located at 10q23-q24, and the LRGI2 gene, located in 1p13.2; nevertheless, in up to 16% of patients, no associated mutations had been found. Osorio et al. [7] also stated the following:
• Recent evidence had suggested that these genes are critical to an adequate neurological development to the lower urinary tract and that the origin of the disease seems to be due to peripheral neuropathy.
• There is clinical variability among patients who are affected by UFS and not all patients do present with the classic two components, and it had even been genetically confirmed in patients with a previous diagnosis of Hinman Syndrome or other urinary bladder dysfunctions.
• Also, the presence of nocturnal lagophthalmos in these patients had been recently reported.
• Early recognition and timely diagnosis of Ochoa syndrome are critical for the prevention of complications such as urinary tract infections or chronic kidney disease.
Aydogdu et al. [15] iterated that the urofacial syndrome, which is also referred as Ochoa syndrome, is a rare autosomal recessive condition which occurs in both genders and characterised by uropathy and facial abnormalities. They also stated that early diagnosis of Ochoa syndrome is crucial for the management and prognosis of urinary problems due to a dysfunctional bladder. Aydogdu et al. [15] reported 11 patients who had urofacial syndrome in five families from Turkey with a median follow up of 32 months and the reported ages of the patients had ranged between 2 months and 44 months.
Elejalde et al. [1] had reported seven patients (4 females, and 3 males) who were born in unrelated families, one of them consanguineous (first cousins), who were afflicted by peculiar facies and gestures while smiling and crying, and by hydronephrosis, hydroureter and intravesical stenosis of the ureter, abnormal calibre of the urethra within the prostatic and membranous portions of the urethra, urethral valves, abnormal urinary bladder with trabeculation, and diverticula associated with severe hypertrophy of the mucosa with sclerotic changes. Elejalde et al. [1] stated that the genetic analysis of these families had suggested that the condition is probably autosomal dominant, with variable expressivity and incomplete penetrance. Elejalde et al. [1] also iterated that the syndrome represents alteration of facial and urinary developmental fields as well as they stated that the peculiar facies, allows early recognition of the condition, and this could be helpful for early assessment and treatment, leading perhaps to a better prognosis.
Wang et al. [3] iterated that the urofacial (Ochoa) syndrome (UFS) is a rare autosomal recessive disease which is typified by congenital obstructive uropathy and abnormal facial expression. They also stated that the patients do present with enuresis, urinary-tract infection, hydronephrosis, and voiding dysfunctions as a result of neurogenic bladders. Elejalde et al. [3] reported that in order to map the UFS gene, a genome screen utilising a combination of homozygosity-mapping and DNA-pooling strategies was undertaken in 20 selected patients, one patient pool, and three control pools (unaffected relatives). With regard to the results of their study, Wang et al. [3] reported the ensuing:
• Pursuant to the analyses of 36 randomly chosen markers, D10S677 was identified as being linked to and associated with UFS, as was suggested by a significant excess of homozygosity in patients compared with that in unaffected relatives (P < 10>• Ten additional markers flanking D10S677 and covering a 22-cM region then were analysed to fine-map the UFS gene by use of haplotype (linkage disequilibrium) analysis.
• All 31 patients had been found to be homozygous for two closely linked markers (D10S1726 and D10S198) located approximately 5 cM telomeric to D10S677, whereas only 12% of the unaffected relatives were homozygous for both markers (P < 10>• Several patients were found to be heterozygous at two markers immediately flanking D10S1726/D10S198, one on the centromeric side (D10S1433) and the other on the telomeric side (D10S603).
• These recombinational events had placed the UFS gene near D10S1726/D10S198 and within a 1-cM interval defined by D10S1433 and D10S603 on chromosome 10q23-q24.
Al-Qahtani et al. [16] iterated that Ochoa syndrome is an uncommon autosomal recessive disease which is typified by congenital obstructive uropathy and abnormal facial expression, as well as they documented that the presence of the latter does enable early recognition and urological evaluation with institution of prophylactic measures thereby preventing further damage to the upper urinary tract. Al-Qahtani et al. [16] had reported three Saudi Arabia siblings who had presented with inverted facial expression, voiding dysfunction, and variable degrees of renal insufficiency with the presence of variable pathology in gall bladder.
garcia-minaur et al. [17] reported on three european cases of urofacial (ochoa) syndrome. garcia-minaur et al. [17] iterated the ensuing:
• This clinical entity was originally described in Colombian patients and very few cases had been reported from other countries.
• It is likely that Ochoa syndrome may be missed because of variability of the urinary problems and failure to recognize the characteristic facial grimacing.
• Establishing an early diagnosis does have important consequences with regard to the management and prognosis of urinary problems in these patients
Pang et al. [18] iterated that previously, they had localized the defective gene for the urofacial syndrome (UFS) to a region upon chromosome 10q24 by homozygosity mapping. Pang et al. [18] reported evidence that Heparanse 2 (HPSE2) is the culprit gene for the syndrome. Pang et al. [18] also reported that mutations with a loss of function in the Heparanase 2 (HPSE2) gene were found in all UFS patients originating from Colombia, the United States, and France. They also iterated that HPSE2 encodes a 592 aa protein that contains a domain showing sequence homology to the glycosyl hydrolase motif in the heparanase (HPSE) gene, but its exact biological function had not yet been characterized. Complete loss of HPSE2 function in UFS patients had indicated that HPSE2 might be important for the synergic action of muscles that are implicated in facial expression and urine voiding.
Chauve et al. [10] stated the ensuing:
• The urofacial syndrome (UFS) or Ochoa syndrome had been reported as a rare autosomal recessive disorder which comprises of a uropathy and facial abnormalities.
• The gene was mapped on chromosome region 10q23-q24.
Chauve et al. [10] had reported the first European cases of UFS. They reported that haplotype analyses in their French family were compared with those that had been previously described in patients from Columbia and America (literature data). They also reported that their results were compatible with the same localization of the critical region and they did favour the postulate of genetic homogeneity.
Ganesan et al. [19] iterated that the Ochoa syndrome is the association of a non-neurogenic neurogenic urinary bladder with abnormal facial muscle expression and that patients are at risk for the development of kidney failure due to obstructive uropathy. Ganesan et al. [19] reported a family of three siblings, with an emphasis on the abnormalities in facial expression. Ganesan et al. [19] iterated the ensuing:
• Careful examination does demonstrate an unusual co-contraction of the orbicularis oculi and orbicularis oris muscles only when full facial expressions are exhibited, across a range of emotional or voluntary situations. This indicates a peripheral disorder in facial muscle control.
• Two thirds of patients who have Ochoa syndrome do tend to have anal sphincter abnormalities. Aberrant organisation of the facial motor and urinary-anal sphincter nuclei might explain these symptoms.
Cesur Baltacı et al. [20] iterated the ensuing:
• Ochoa syndrome (UFS1; Urofacial syndrome-1) is a very uncommon autosomal recessive disorder that is caused by mutations in the HPSE2 gene which emanates in urinary bladder voiding dysfunction and somatic motor neuropathy affecting the VIIth cranial nerve.
• Niemann-Pick disease is a rare autosomal recessive lysosomal storage disorder that is associated with systemic involvement resulting from sphingomyelinase deficiency and generally occurs via mutation in the sphingomyelin phosphodiesterase-1 gene (SMPD1).
Cesur Baltacı et al. [20] had reported a 6-year-old girl who had symptoms such as urinary incontinence, recurrent urinary tract infections, peculiar facial expression, mainly when smiling, hypertelorism, constipation, incomplete closure of eyelids during sleep and splenomegaly. Homozygote mutations in two different genes responsible for two distinct syndromes were identified in the patient. Cesur Baltacı et al. [20] had also reported that homozygous NM_000543.5:c.502G>A (p.Gly168Arg) mutation was identified in the SMPD1 gene causing Niemann-Pick disease. Cesur Baltacı et al. [20] also iterated that in addition, some of the clinical features were due to a novel homozygous mutation identified in the HPSE2 gene, NM_021828.5:c.755delA (p.Lys252SerfsTer23). Cesur Baltacı et al. [20] made the following conclusions:
• They had discussed about the importance of considering dual diagnosis in societies where consanguineous marriages are common.
• Accurate diagnosis of the patient is very important for the management of the diseases and prevention of complications.
Wang et al. [21] iterated the ensuing:
• The urofacial (Ochoa) syndrome (UFS) is typified by congenital obstructive uropathy and abnormal facial expression is an uncommon disorder which is caused by a single recessive disease gene.
• Their previous studies using homozygosity mapping had located the UFS gene to a genomic interval of approximately 360 kb on chromosome 10q23-10q24.
• In their study, they had constructed a genomic sequence map which had covered the entire UFS interval and narrowed the disease interval to a genomic region of 220 kb which harbour the newly identified ACDP1 gene in addition to part of the GOT1 gene which had already been excluded as a candidate for UFS.
• Their extensive search for mutations within the coding region, the 5′ and 3′ untranslated regions, the promoter region, and the exon/intron junctions had failed to find a pathogenic mutation in UFS patients.
• In addition, their analyses had indicated that the same gene on chromosome 10q is responsible for all UFS patients from multiple ethnic groups.
Derbent et al. [22] iterated the ensuing:
• Urofacial (Ochoa) syndrome is an uncommon autosomal-recessive disorder that features an unusual “inverted” facial expression, such that patients appear to be crying when they smile.
• This syndrome also entails serious urinary tract disorders, even though the diagnosis may be missed because of variability of these problems and failure to recognize the characteristic facial grimacing.
• The urinary issues usually emanate in the development of enuresis, urinary tract infection, and hydronephrosis, and some severely affected patients do become hypertensive and progress to end-stage renal disease.
• Early diagnosis is very important for the management of urinary problems and best prognosis in these patients.
Derbent et al. [22] reported the first published case of urofacial syndrome in Turkey. The patient was diagnosed at 16 years of age, after having been followed-up with the diagnosis of recurrent urinary tract infection and vesical-ureteral reflux. Derbent et al. [22] recommended that physicians should keep this syndrome in mind for any patient who presents with dysfunctional voiding, particularly in countries with high rates of consanguineous marriage.
Barbon et al. [23] iterated the ensuing:
• The Urofacial or Ochoa Syndrome is a very uncommon congenital disorder which includes vesical bladder dysfunction and a peculiar inverse facial expression, that brings patients to express a sad-crying face while they intend to laugh.
• Up-to-date treatments had addressed only the urological side of this disease. However, also the impaired facial mimicry does have a strong impact upon patients' quality of life.
Barbon et al. [23] treated a young patient with Botulinum toxin in order to address this impairment and obtained pleasing results, including a harmonic smile and a very satisfied patient. Barbon et al. [23] also stated the following:
• To the best of their knowledge, their reported case was the first time that the use of Botulinum toxin had been reported in literature to address the facial expression component of this disease.
Stuart et al. [24] iterated that Urofacial syndrome (UFS) (or Ochoa syndrome) is an autosomal-recessive disease which is typified by congenital urinary bladder dysfunction, which is associated with a significant risk of renal failure, and an abnormal facial expression upon smiling, laughing, and crying. Stuart et al. [24] reported that a subset of UFS-affected individuals, do have biallelic mutations in LRIG2, encoding leucine-rich repeats and immunoglobulin-like domains 2, a protein implicated in neural cell signalling and tumorigenesis. Stuart et al. [24] iterated that importantly, they had demonstrated that rare variants in LRIG2 may be relevant to non-syndromic bladder disease. Stuart et al. [24] also documented that they had previously demonstrated that UFS is also caused by mutations in HPSE2, encoding heparanase-2. LRIG2 and heparanase-2 were immunodetected in nerve fascicles growing between muscle bundles within the human foetal bladder, directly implicating both molecules in neural development in the lower urinary tract.
Wolf et al. [25] documented the ensuing summations related to Ochoa syndrome:
• The urofacial or Ochoa, syndrome is characterised by congenital urinary dysfunction together with an abnormal grimace upon smiling, laughing or crying.
• Ochoa syndrome could present as foetal megacystitis as a result of simultaneous detrusor muscle and urinary bladder outlet contractions.
• Vesical-ureteric reflux often tends to be manifest in Ochoa syndrome and the condition could be complicated by urosepsis and end-stage renal disease.
• Ochoa syndrome had been postulated to be associated with neural basis, and it could be familial when it is inherited in an autosomal recessive fashion.
• Majority of individuals who had urofacial syndrome that had been reported up to the time of publication of their article, had carried biallelic, postulated functionally null mutations of HPSE2 or, less commonly, LRIG2.
• Little is known about the biology of the respective encoded proteins, heparinase 2 and leucine rich repeats and immunoglobulin-like domains 2.
• However, the observation that heparinase 2, could bind heparan sulphate proteoglycans as well as inhibit heparinase 1 enzymatic activity and that LRIG2 could modulate receptor tyrosine kinase growth factor signalling each point to biological roles that are relevant to tissue differentiation.
• In addition, both heparinase 2 and LRIG2 proteins are identified in automatic nerves growing into foetal urinary bladders.
• The collective evidence had been consistent with the postulate that urofacial syndrome genes do encode for proteins that work in a common pathway in order to facilitate neural growth into, and / or function within, the urinary bladder.
• This molecular pathway may also have relevance to the understanding of the pathogenesis of other lower tract diseases, including Hinman-Allen syndrome, or non-neurogenic neurogenic urinary bladder, and the subset of individuals who have primary vesical-ureteric reflux accompanied by urinary bladder dysfunction.
Newman et al. [26] made the ensuing summating iterations related to various aspects of Ochoa Syndrome as follows:
Clinical characteristics:
• Urofacial syndrome (UFS) is characterised by prenatal or infantile commencement of urinary bladder voiding dysfunction, abnormal facial movement with [removed]resulting from abnormal co-contraction of the corners of the mouth and eyes), and often bowel dysfunction (constipation and/or encopresis).
• Urinary bladder voiding dysfunction does increase the risk for urinary incontinence, megacystis, vesicoureteric reflux, hydroureteronephrosis, urosepsis, and progressive renal impairment.
• In rare instances, an individual who has (a) a molecularly confirmed diagnosis and/or (b) an affected relative meeting clinical diagnostic criteria presents only the characteristic facial features or only the urinary bladder voiding dysfunction (not both) would be diagnosed with Ochoa syndrome.
• Nocturnal lagophthalmos (incomplete closing of the eyes during sleep) appears to be a common and significant finding in Ochoa syndrome.
.Diagnosis/testing:
• The diagnosis of Ochoa Syndrome (UFS) has been based upon investigations of the urinary tract which demonstrate characteristic urinary tract abnormalities as well as physical examination which reveals characteristic facial movement with expression.
• Ochoa Syndrome / Urofacial Syndrome (UFS) is a heterogeneous condition developing from biallelic pathogenic variants in either HPSE2 or LRIG2.
• In some instances, no pathogenic change had been found in Ochoa syndrome.
• It is worth noting that the majority of individuals who have UFS that had been reported to date had not had molecular confirmation of their diagnosis undertaken by their clinicians.
Management:
The ensuing educative summations could made about the management of Ochoa syndrome:
• Treatment of manifestations:
Rapid and complete treatment of urinary tract infections and routine treatment of urosepsis should be undertaken.
With regard to urinary incontinence and urinary bladder dysfunction: utilization of anticholinergic and α1-adrenergic blockers; intermittent self- urethral catheterization or vesicostomy should be undertaken.
Surgical management of hydroureteronephrosis (which in this case could entail the insertion of percutaneous nephrostomy or insertion of a double J ureteric stent) and urinary bladder augmentation should be considered.
Management of chronic kidney disease and end-stage renal disease relies upon the standard optimal options.
• Surveillance:
Monitoring for evidence of urinary tract features including vesical-ureteric reflux and hydroureteronephrosis should be undertaken.
Renal function should be monitored at intervals which should be determined by urinary tract features at manifestation and their subsequent progression.
• Agents/circumstances to avoid:
Nephrotoxic substances.
• Evaluation of relatives at risk:
It is appropriate to examine siblings of an affected individual as soon as possible after birth in order to ascertain if facial and / or urinary tract manifestations of UFS are present in order to enable prompt evaluation of the urinary tract and renal function and prompt initiation of necessary treatment. (It important to assess individuals for constipation problems as well as inability to close eye lids at night or during sleeping which was not stated by the authors in their summation).
• Pregnancy management:
Although no guidelines for prenatal management of UFS does exist, it does seem appropriate to undertake ultrasound scan examination of pregnancies at risk in order to ascertain if urinary tract involvement of UFS is present, as this might influence the timing and / or location of delivery (e.g., in a tertiary medical centre which could manage renal/urinary complications immediately after birth).
• Genetic counselling:
UFS is inherited in an autosomal recessive manner.
At conception, each sibling of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
If the pathogenic variants in the family are known, carrier testing for at-risk relatives and prenatal and preimplantation genetic testing are possible.
Skálová et al. [27] iterated the ensuing:
• The urofacial (Ochoa) syndrome (UFS) is a very rare autosomal recessive disorder which is characterized by abnormal facial expression and urinary abnormalities.
• Patients who are afflicted with this syndrome have urinary tract infection, hydronephrosis, hydroureter and voiding dysfunctions resulting from neurogenic bladder, together with a peculiar inverted facial expression, mainly when smiling or crying.
• This syndrome had been so far observed in Colombia, USA, France and Spain.
Skálová et al. [27] had reported the first case of Ochoa syndrome in the central European population. Skálová et al. [27] documented the ensuing:
• The urofacial (Ochoa) syndrome (UFS) is a very uncommon autosomal recessive disorder which is characterized by abnormal facial expression and urinary abnormalities.
• Patients who are afflicted by this syndrome have urinary tract infection, hydronephrosis, hydroureter and voiding dysfunctions resulting from neurogenic bladder, together with a peculiar inverted facial expression, mainly when smiling or crying.
Skálová et al. [27] reported a patient, who was a girl of young unrelated parents of Czech origin. The father of the girl had been treated for Crohn’s disease, while her mother and the patient’s brother, who was 5 years of age at that time, were healthy. Her prenatal ultrasound scan was normal in the 20th week of pregnancy; however, later on, within the 35th week of gestation, hypotrophy and megavesica with bilateral hydronephrosis were demonstrated upon the ultrasound scan. Therefore, the child was delivered on the 36th week of pregnancy by a Caesarean section. During the course of her delivery, rupture of the urinary bladder occurred, which was consequently treated by the bladder suture. There were no signs of abdominal wall abnormalities. The girl’s birth weight was 1760 grams. Her postnatal period was complicated by recurrent sepsis and hypertension, both of which were successfully treated. Abdominal ultrasound scan was undertaken which demonstrated dilated calices and a large neurogenic bladder. There were no signs of vesicoureteral reflux on voiding cystourethrography. Infra-vesical obstruction was not demonstrated. In view of the urinary retention, Blocksom’s vesicostomy was undertaken at three months of age. Bizarre facial expression was identified when the child was crying and this was even more apparent after the 3rd month of age when the girl attempted to smile (see figure 1). Furthermore, recurrent septic states had occurred within the first 3 months of her age, and this was attributed to a transient hypogammaglobulinaemia. The girl was discharged at the age of 5 months and she was continuously followed-up, receiving prophylactic regimen with co-trimoxazole or nitrofurantoin, and repeatedly hospitalised for recurrent pyelonephritis which was treated with cefuroxime, amoxicilline/ clavulonate, gentamicine and cefixime, respectively. Due to hypogammaglobulinaemia, intravenous immunoglobuline was periodically administered until 18 months of age. Her urinary tract dilation of the girl gradually progressed and her facial inversion became even more apparent. At the age of 25 months the child was admitted to the department of the authors, in view of several hours lasting history of fever and vomiting. Urosepsis was the diagnosis and this rapidly progressed to septic shock, and in spite of antibiotic therapy and re-animation the child died within 24 hours following her admission.
Skálová et al. [27] made the ensuing summating discussions:
• The UFS had been described by Professor. Bernardo Ochoa in a Colombian population. [1] [2] [14] [28]
• As was iterated above, the typical signs of UFS are urinary tract infection, hydronephrosis, hydroureter and voiding dysfunctions resulting from neurogenic bladder, together with a peculiar inverted facial expression, mainly when smiling or crying.
• About two thirds of the patients also manifest with moderate to severe constipation due to bowel dysfunction. [2] [27] [28]
• The simultaneous involvement of the urinary bladder and facial muscles stems from the fact, that normal micturition is a brain stem reflex which is centred within the reticular formation where it has a close anatomical relationship to the origin of facial nerves.
• As the centre for micturition (the pontine micturition centre) and closely related pontine urine storage centre are located at the reticular formation [29] These share the same topographic location with the so-called laughing and crying centre which is located above the facial and respiratory nuclei, most probably in the upper pons or midbrain [30]
• Lesions within this area could conceivably produce dyssynergia manifested in a variety of organ systems [14] [28]
• Understanding the three complementary but different components of the UFS is essential for the correct diagnosis and management of these children.
• The three components of Ochoa syndrome are the genetic background, the dysfunction of the facial expression, and the dysfunctional emptying of urine and faeces [14]
• This most unusual disorder had been initially considered to be a local observation, as majority of patients with UFS came from Colombia [1] [14] [27]
• However, children who have UFS had been also later reported from various countries, in particular in Americans of Irish descent [4] [7]. and in two Arabic children [31] [32].
• The first reported European cases of Ochoa syndrome were from France [10] and Spain [17] [33]
• In a recent report from Canada, 3 patients of Caucasian origin were diagnosed as having UFS. [34] The haplotype analyses in the French family [9] were adjudged to be compatible with those that had been described in Colombian and American-Irish families [3] [4] [35]
• The UFS gene had been mapped to chromosome 10q23–q24 [3] [4] [32] [35] [36] and was reported as responsible for all UFS patients from various ethnic groups. [36]
• The occurrence of the disorder in multiple siblings with normal parents and increased consanguinity, as well as equal distribution according to sex, support autosomal recessive inheritance [2]. [14] [28]
• It has been stated that the inversion of the facial expression which characterises the UFS is not a structural facial defect, but a dysfunctional expression [14]
• The facial identity in patients who have UFS is similar to normal individuals, but not the facial expression when they laugh.
• It has been pointed out that when the patients who have UFS are at rest or when they are sad or suffer pain or when they cry the facial expression is the same as in normal persons. Nevertheless, when they laugh, they grimace as if they are expressing sadness, discomfort or pain [14].
• Urinary bladder dysfunction syndrome, voiding dysfunction syndrome, elimination dysfunction syndrome, non-neurogenic neurogenic urinary bladder and urofacial syndrome all do have the typifying spectrum of symptoms and signs of the neurogenic or obstructive bladder, without apparent neurological or obstructive disease [14].
• Dysfunctional elimination and dysfunctional voiding, is an aggressive disease, as the patients proceed to serious renal deterioration. It has been pointed out that the management of these patients essentially does not differ from that used in the treatment of other voiding disorders of neurological origin [1] [2] [14] [28]
• The principal goal in the management of Ochoa syndrome is to prevent irreversible renal failure. The presence of urinary tract abnormalities and bizarre facial expression in our patient suggest the diagnosis of Ochoa syndrome.
• The recurrent urinary tract infections need to be attributed to the urinary tract malformation; nevertheless, transient hypogammaglobulinaemia might have influenced this as well.
• The combination of urinary tract abnormalities and hypogammaglobulinaemia could have contributed to the unfavourable outcome of their patient.
• To their knowledge, their reported case, was the first reported case of UFS in a Central European population.
• The occurrence of UFS could be expected in various unrelated populations.
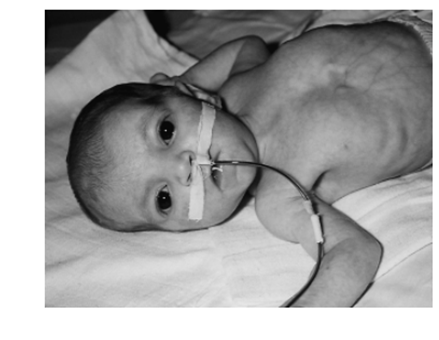
Figure 1. – Peculiar Facial expression Patient’s attempt to smile. The Urofacial syndrome. Reproduced from: [27] Under the Creative Commons Attribution License.

Figure 2: Patient with the inverted face sign that is pathognomonic of Ochoa Syndrome. Reproduced from: [38] Under the Creative Commons Attribution License.

Figure 3: Nocturnal lagophthalmos incomplete closure of the left eye during sleep.
Reproduced from: [38] Under the Creative Commons Attribution License.
The patient’s diagnosis was a clinical diagnosis, since she had manifested with the pathognomonic sign of the disease which had comprised of: the inverted facial expression or inverted smile, characterised by the facial manifestation of crying when the individual with the pathology smiles; The authors had pointed out that it should be realised that at the time of her initial diagnosis, about 15 years preceding the publication of the article, in view of herself and her family’s socioeconomic condition, genetic tests were not available.
She had 8 siblings, 1 male, 7 females (1 half-sister on her mother’s side) (Figure 3). Of these, four cases of Urofacial Syndrome (Ochoa’s) had already been diagnosed in the family, including the patient, who was diagnosed at the age of 7 years, two sisters who were diagnosed since birth, and the half-sister who, even though she was older, for being practically asymptomatic from the urological point of view, the diagnosis was confirmed only after the patient. The patient denied knowledge about parental consanguinity (both parents had already been deceased).
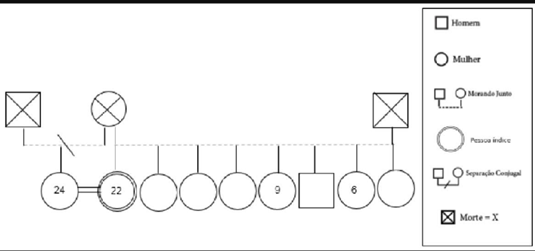
Figure 4: Genogram of the patient.
In addition, the patient had continued having with joint follow-up from nephrology and urology to verify the feasibility of kidney transplantation, with or without the need for prior bladder enlargement. The patient was undergoing tests and follow-ups to assess the viability of a kidney transplant. Reproduced from: [38] Under the Creative Commons Attribution License.
Barros Jacobino et al. [38] made the ensuing detailed summating discussions:
• It has been iterated that Ochoa syndrome was first described by Bernardo Ochoa in the 1960s, and the inverted facial smile does enable the early identification of this syndrome and precedes the appearance of urological symptoms. [14]
• Age might or might not correlate with the severity of the symptoms of the disease, as the pathological findings might go unnoticed on prenatal ultrasounds scans. Hence, patients might have severely compromised renal function from birth or after a few years.
• However, it had been pointed out that it is well known that the delay in the diagnosis of Ochoa Syndrome might culminate in the development of greater renal repercussions, proving the importance of early diagnosis [7]
• The two classic components of Ochoa Syndrome (the characteristic smile and impaired urinary bladder emptying) in patients who are affected by Ochoa Syndrome usually tend to be manifest in individuals who have this pathology (see Chart 1).
• Despite the variability of presentations, the urinary bladder dysfunction follows the same biological behaviour in all clinical manifestations of Ochoa Syndrome.

Reproduced from: [38] Under the Creative Commons Attribution License.
• It has been documented that clinical cases do bring in their presentations with signs and symptoms that might be present in each patient, such as craniofacial abnormalities typified by an inverted smile [39] [40]. urinary bladder dysfunction. [41] [42].and nocturnal lagophthalmos [6]
• Ochoa Syndrome was one of the uncommon cases reported in the state of Piauí, with a patient presenting with the complete characteristics of the inverted face pathology and complications in the urinary tract. Their reported case had simulated the case which was reported in another study, regarding the characteristic manifestation of peculiar facial expressions and chronic renal failure and associated complications [37]
• It has been documented that the inverse facial expression has functional characteristics, being a pathognomonic finding of the disease, not finding explanations of morphology alterations or injuries [41]
• In one reported study, it was found that for 25 families who were afflicted by the disease, 21 of them had a mutation within one of the two genes associated with the disease, and 15% might not have any of the affected genes [43]
• Mutation of the HPSE2 gene, mapped to chromosome 10q23-q24 which encodes the heparanase 2 protein or the LRIG2 gene which encodes the protein that promotes epidermal growth factor signalling, was found to be related to aberrant bladder innervations.
• In addition, it had been iterated that these genes act upon the facial nerve, which is closely related to the inverted smile, which is, the characteristic grimace when smiling [44]
• Mutations within the HPSE2 gene had already been studied in mice, which had confirmed the relationship with urinary bladder dysfunction, with excessive formation of fibrotic tissue from the accumulation of collagen deposits, emanating in organ remodelling, more common in the detrusor. [45].
• In another study which had been undertaken in mice involving the presence or absence of homozygous mutations in the study group, when evaluating the immunohistochemistry, a prevalence of nerve fibres towards the detrusor and a decrease in those directed to the urinary bladder outflow tract were found, common findings in the bladder. hypercontractile, which is how it appears in Urofacial Syndrome [46]
• Patients who have Urofacial or Ochoa Syndrome almost always do have alterations in urinary emptying due to sphincter-detrusor dyssynergia, consequently, high bladder pressures and/or post-voiding residues do occur which promote vesicular reflux, hydronephrosis, UTI and renal scarring with subsequent development of CKD. In addition, a trabeculated urinary bladder with thickened walls could be found [47].
• As for the findings of lagophthalmos, it is followed by persistent ocular symptoms and exposure to keratopathy [48]
• It could be a clinical characteristic that is seen by parents, guardians, or people who live with it for a long time, but it could also be manifested in such a light way, not being found or found again. For this reason, its true frequency in patients with the syndrome was still not known [7]
• There is a high likelihood that the patient would progress to terminal chronic kidney disease (CKD), mainly as a result of late diagnosis, and it is important to note that there are few cases of successful transplants in these patients, as there is an increased risk of post-transplant urinary tract infection) UTI [35] [48]
• In less severe scenarios, urinary bladder enlargement had been recommended, either with or without continent external urinary diversion.
• The option of intermittent urinary bladder catheterization is recommended when the objective is to prevent damage to the renal graft. [48]
Barros Jacobino et al. [38] made the ensuing conclusions:
• One of the causes of the rarity of this syndrome is related to the lack of knowledge upon the part of the medical community, neglecting its diagnosis and the consequent institution of adequate early treatment.
• In view of this, it is important to evaluate each case and, in case of clinical suspicion, it is important to undertake genetic research, when available, including close family members, for proper follow-up, in order to prevent the disease from developing into serious complications, such as end-stage chronic kidney disease.
• Therefore, it is evident that early diagnosis with the undertaking of adequate treatment, avoids possible damage to the urinary tract from childhood, which enables better management and quality of life in these patients.
Govindarajan et al. [49] iterated that Ochoa or urofacial syndrome is a rare autosomal recessive syndrome with around 150 cases reported in the medical literature which had entailed neurogenic bladder and facial abnormalities, culminating in obstructive uropathy and chronic kidney disease. Govindarajan et al. [49] reported a 5-year-old boy, who had manifested to their establishment with Stage IV chronic kidney disease with bilateral hydroureteronephrosis secondary to chronic urinary incontinence. His peculiar facial expression with a grimace while smiling had indicated the diagnosis of Ochoa syndrome. He was managed conservatively for neurogenic bladder and he was under follow-up at the time of the report of the case. Govindarajan et al. [49] highlighted this unique syndrome and the simplicity in making this syndromic diagnosis, just by the appreciation of abnormal facial expressions.
Hazzab et al. [50] reported a 6-year-old girl who was the second child of consanguineous parents (uncle ± niece). Her father was reported to have suffered episodes of urinary tract infection. He did not demonstrate the characteristic grimacing of his daughter. This child was born at full-term following a normal vaginal delivery pursuant to an uneventful pregnancy. Her birth weight was 2500 grams and length 49.5 cm. Her development was otherwise normal in the first years of her life, but she had suffered from enuresis. At the age of 6 years, she had manifested with urinary tract infection. The results of her serum urea and creatinine were increased and her GFR was 20 ml/min; her renal echography demonstrated enlarged-kidneys, with ureteral, and calyceal dilatation, and a low-compliance bladder with pseudo-diverticular, and a significant residual urine after voiding: 130 ml. She had voiding cystourethrography which had revealed a trabeculated urinary bladder and a significant post-micturition residue (see Figure 5). This child had demonstrated the characteristic, inverted, facial [removed]see Figure 6).

Figure 5: Uretherocystographie Reproduced from: [50] Under the Creative Commons Attribution License.
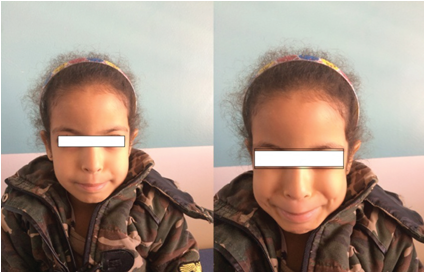
Figure 6: Facial expression of Ochoa Syndrome: Reproduced from: [50] Under Creative Commons Attribution License.
Based upon the suspicion of urinary bladder dysfunction urodynamics studies were undertaken. The urodynamic exploration demonstrated features of a non-neurogenic bladder. Adding the urodynamic findings of a non-neurogenic bladder to the typical facial expression, the diagnosis of Ochoa Syndrome was established. Genetic analysis was not undertaken due to unavailability of Genetic analysis within Morocco and the high cost of genetic analysis. She was initially treated with antibiotic prophylaxis and anticholinergics and conservative treatment of her renal failure as well as with clean intermittent catheterization to empty her urinary bladder.
Hazzab et al. [50] made the ensuing summating discussions:
• Urofacial syndrome (UFS) is typified by prenatal or infantile onset of urinary bladder voiding dysfunction, abnormal facial movement with [removed]resulting from abnormal co-contraction of the corners of the mouth and eyes), and often bowel dysfunction (constipation and/or encopresis).
• Urinary bladder voiding dysfunction does increase the risk for the development of urinary incontinence, megacystis, vesical-ureteric reflux, hydroureteronephrosis, urosepsis, and progressive renal impairment.
• In rare instances, an individual who has a molecularly confirmed diagnosis of Ochoa Syndrome and/or an affected relative meeting clinical diagnostic criteria manifests with only the typifying facial features or only the urinary bladder voiding dysfunction (not both).
• Nocturnal lagophthalmos (incomplete closing of the eyes during sleep) does appear to be a common and significant finding in Ochoa syndrome [51]
• UFS was first reported in many unrelated Colombian families (Elejalde, 1979; Ochoa and Gorlin, 1987). [1] [2]
• The facial abnormality is a typifying 'inverse' facial expression when these patients smile, such that they do appear to be crying.
• This grimacing is highly diagnostic of the UFS.
• Ochoa (1992) had studied 50 patients from 32 families, the largest series which had been reported so far. The majority of the patients were reported to have manifested with enuresis with or without urinary tract infection. A third of these patients had been referred because of the typifying facial expression, and the remainder had been referred because of urinary tract infection. Constipation was noted in more than half of the patients which amounted to 28 patients out of 50 patients. [28]
• Urological investigations and urodynamic studies of the patients had demonstrated characteristic findings of neuropathic bladder in all of the patients which had included: A trabeculated, large capacity urinary bladder with narrowing of the urethral lumen in all of them; a vesicoureteral reflux in two-thirds of patients which amounted to 32 out of 50 patients, which was bilateral in half of them that amounted to 18 out of 32; and a hypertonic, hyper reflexic type of urinary bladder in about half of the patients that amounted to 22 out of 50 patients. None of the parents had demonstrated the characteristic facial expression, or any abnormal finding in their urological investigations.
• The observation of a large number of UFS cases which are confined to a small geographical area could support a founder effect. However, further cases had been reported from Kuwait (Teebi and Hassoon, 1991), [32] and the United States (Wang, et al. 1999) [4], as well as from Europe Galan, et al. 1997; and Chauve, et al. 2000). [10] In addition, genetic homogeneity had been demonstrated in the reported North American and French families (Wang, et al. 1999; [4] Chauve, et al. 2000) [10]. Even though undoubtedly a rare condition, it seems possible that there might be cases of UFS in whom the diagnosis had not been made in view of the absence of overt urinary tract symptoms [17]
• UFS is a heterogeneous condition resulting from biallelic pathogenic variants in either HPSE2 or LRIG2.
• In some instances, no pathogenic change had been found.
• It is worth noting that the majority of individuals who have UFS reported up to the time of publication of their article, had not had molecular confirmation of their diagnosis.
• Ochoa Syndrome is inherited in an autosomal recessive manner. At conception, each sibling of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives, prenatal testing of pregnancies at increased risk, and preimplantation genetic diagnosis should be possible if the pathogenic variants in the family are known [51]
• The aim of early treatment of Ochoa Syndrome entails the restoration of balanced bladder emptying and prevention of upper tract deterioration. By achieving a low-pressure and adequate urinary bladder capacity, the potential risk of upper urinary tract dilation, associated reflux, thus, progressive renal scarring could be eliminated. Significant residual urine could cause recurrent febrile urinary tract infections. Because of this, CIC is inevitable in majority of the patients. Therapeutic strategies entail provision of prophylactic antibiotics, clean intermittent catheterization, anticholinergics, and intravesical botulinus toxin injection, if required. Efficacy of the treatment has varied between individuals who have Ochoa syndrome (see Table 2) [15]
• Urological problems, radiological findings, and management of Ochoa syndrome patients.[50]
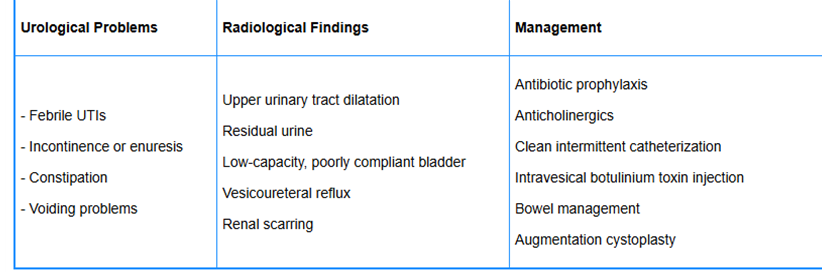
Table 2: Urological problems, radiological findings, and management of Ochoa Syndrome Patients. Reproduced from: [50] Under Creative Commons Attribution License.
Surveillance
• Monitor:
o For evidence of urinary tract features including vesical-ureteric reflux and hydroureteronephrosis;
o Renal function at intervals determined by urinary tract features at presentation and their subsequent progression;
o For evidence of significant corneal involvement in individuals with nocturnal lagophthalmos.
• Agents/Circumstances to avoid:
o Nephrotoxic substances are contraindicated in individuals who have renal impairment and nephrotoxic substances should be avoided if possible.
• Evaluation of relatives at risk:
o It is appropriate to clarify the genetic/clinical status of sibs of an affected individual as soon as possible after birth in order to identify those who would benefit from prompt evaluation of the urinary tract and renal function and early initiation of necessary treatment.
• Evaluations can include:
o Molecular genetic testing if the pathogenic variants in the family are known;
o Examination to determine whether facial and/or urinary tract manifestations of UFS are present if the pathogenic variants in the family are not known.
o Genetic Counselling for issues related to testing of at-risk relatives for genetic counselling purposes should be undertaken.
• Pregnancy management:
o Even though no guidelines for prenatal management of UFS exist, it seems appropriate to perform ultrasound examination of pregnancies at risk to determine if urinary tract involvement of UFS is present, as it may influence the timing and/or location of delivery (e.g., in a tertiary medical centre that could manage renal/urinary complications immediately after birth).[51]
Hazzab et al. [50] made the following conclusions:
• Ochoa syndrome should always be considered in patients who manifest with dysfunctional urinary bladder who have characteristic grimacing when smiling.
• The prognosis of urofacial syndrome is generally poor and does require multiple treatment modalities.
• Early diagnosis of the syndrome is mandatory.
Mermerkaya et al [6] stated the following;
• The urofacial syndrome is a rare condition that afflicts both genders and typified by uropathy and facial abnormalities.
• Early diagnosis is crucial for the management and prognosis of urinary problems.
• Paradoxical inversion of facial musculature when smiling, giving an appearance of crying associated with severe urinary tract dysfunction is typical in these patients who are afflicted by urofacial syndrome.
• Even though facial signs and symptoms had generally tended to be ignored and shadowed by the dominant urinary bladder symptoms, they had recently realized a unique but constant finding in the majority of these patients, nocturnal lagophthalmos which is described as inability to close the eyelids during sleep.
Mermerkaya et al [6] reported 15 patients with urofacial syndrome (Ochoa) whom mostly had admitted been with major urological symptoms and 12 of the cases had nocturnal lagophthalmos. Mermerkaya et al [6] made the ensuing educative discussions:
• Lagophthalmos might lead to keratitis, corneal abrasion, infection, vascularization, and in extreme cases, ocular perforation, endophthalmitis and loss of the eye.
• Basic modalities like lubricant drops during the day and ointments at night are usually enough to protect the cornea from exposure keratopathy.
• In moderate to severe cases, overnight taping of the lid or utilisation of a moisture chamber might be necessary.
• The majority of their patients had responded to basic therapy.
Mermerkaya et al [6] made the ensuing conclusions:
• Nocturnal lagophthalmos is a novel symptom described in patients with urofacial syndrome.
• Paediatricians as well as urologists should be careful about this symptom to prevent damage to the eye and quality of life problems.
Grenier et al. [52] iterated the ensuing:
• Urofacial, or Ochoa, syndrome (UFS) is an autosomal recessive disease which features a dyssynergic urinary bladder with detrusor smooth muscle contracting against an un-dilated outflow tract.
• It also features an abnormal grimace.
• Half of individuals who have UFS carry biallelic variants in HPSE2, whereas other rare families carry variants in LRIG2.LRIG2 is immunodetected in pelvic ganglia sending autonomic axons into the bladder. Moreover, Lrig2 mutant mice have abnormal urination and abnormally patterned bladder nerves.
• They had postulated that peripheral neurogenic defects underlie LRIG2-associated bladder dysfunction.
Grenier et al. [52] described a new family with LRIG2-associated UFS and studied Lrig2 homozygous mutant mice with ex vivo physiological analyses. Grenier et al. [52] summated the results as follows:
• The index case manifested antenatally with urinary tract (UT) dilatation, and postnatally had urosepsis and functional bladder outlet obstruction.
• He had the grimace that, together with UT disease, characterizes UFS.
• Even though HPSE2 sequencing was normal, he carried a homozygous, predicted pathogenic, LRIG2 stop variant (c.1939C>T; p.Arg647∗). Lrig2 mutant mice had enlarged bladders.
• Ex vivo physiology experiments had shown neurogenic smooth muscle relaxation defects in the outflow tract, containing the urethra adjoining the bladder, and in detrusor contractility.
• furthermore, there were nuanced differences in physiological outflow tract defects between the sexes.
Grenier et al. [52] made the ensuing conclusions:
• Putting this family in the context of all reported UT disease-associated LRIG2 variants, the full UFS phenotype occurs with biallelic stop or frameshift variants, but missense variants lead to bladder-limited disease.
• Their murine observations support the hypothesis that UFS is a genetic autonomic neuropathy of the bladder affecting outflow tract and bladder body function.
Emir et al. [53] reported a child who had Ochoa Syndrome who also had Wilms tumour. The association of Ochoa Syndrome with malignant tumours of various parts of the body had not been generally reported and perhaps this association had so far been underreported and in view of this clinicians who encounter malignant tumours should try to ascertain if their patients also do have Ochoa Syndrome by carefully assessing the patients.
Del Valle-Peréz et al [56] made the ensuing iterations:
• Urofacial syndrome or Ochoa syndrome (UFS or UFOS) is a rare disease which is typified by inverted facial expression and urinary bladder dysfunction which was described for the first time in Colombia.
• Ochoa syndrome is an autosomal recessive pathology with mutations in the HPSE2 and LRIG2 genes.
• Nevertheless,16% of patients are stated not to have any mutations associated with the syndrome.
• Despite the importance of neurobiology in its pathophysiology, there are no neurological, neuropsychological, or psychological studies in these patients.
Del Valle-Peréz et al. [56] reported a 30-year-old man from Medellín, Colombia, with a significant perinatal history, who was diagnosed with grade 4 hydronephrosis on his first ultrasound scan test. At 4 months of age, he developed symptoms such as hypomimia, lagophthalmos, and recurrent urinary tract infections. He underwent radiology imaging studies, which demonstrated urinary tract dilatation, vesicoureteral reflux, and a double collector system on his left side, which had led to the diagnosis of UFS. Multiple procedures, including vesicostomy, ureterostomy, and enterocystoplasty, were undertaken. At 20 years of age, he attained urinary sphincter control. He had genetic analysis which demonstrated a founder pathogenic variant, c.1516C > T (p.Arg506Ter), in the HPSE2 gene, which produces a truncated protein which lacks 86 amino acids. This variant is classified as pathogenic according to the ClinVar database for UFS. The mutation age is stated to be about 260-360 years, and the two alleles share a 7.2-7.4 Mb IBD segment. Moreover, Del Valle-Peréz et al. [56] detected European local ancestry in the IBD segment, which is consistent with a Spanish introduction. Neurological examination, neuropsychological assessment, and psychological testing revealed no abnormalities, except for high stress levels. Clinical analysis of this patient revealed distorted facial expression and detrusor-sphincter dyssynergia, which are typical of patients with UFS. Genetic analysis demonstrated a pathogenic variant in the HPSE2 gene of European origin and a mutation age of 260-360 years. From a neurological, neuropsychological, and psychological (emotional and personality) perspective, the patient had demonstrated no signs or symptoms of clinical interest.
Ochoa et al. [57] stated the ensuing:
• Autoimmune poly-endocrinopathy-candidiasis-ectodermal-dystrophy (APECED) represents a life-threatening monogenic autoimmune disorder which is primarily caused by biallelic deleterious variants in the autoimmune regulator (AIRE) gene.
Ochoa et al. [57] prospectively evaluated 104 patients who were diagnosed clinically as being afflicted by APECED syndrome and they identified 17 patients that amounted to 16% of the patients from 14 kindreds who had lacked biallelic AIRE variants in exons or flanking intronic regions; 15 of the patients had Puerto Rican ancestry. Through whole-genome sequencing, Ochoa et al. [57] had identified a deep intronic AIRE variant (c.1504-818 G>A) cosegregating with the disease in all 17 patients. Ochoa et al. [57] developed a culture system of AIRE-expressing primary patient monocyte-derived dendritic cells and had demonstrated that c.1504-818 G>A creates a cryptic splice site and activates inclusion of a 109–base pair frame-shifting pseudo-exon. Ochoa et al. [57] also found low-level AIRE expression in patient-derived lymphoblastoid cell lines (LCLs) and had confirmed pseudo-exon inclusion in independent extrathymic AIRE–expressing cell lines. Through protein modelling and transcriptomic analyses of AIRE-transfected human embryonic kidney 293 and thymic epithelial cell 4D6 cells, Ochoa et al. [57] showed that this variant alters the carboxyl terminus of the AIRE protein, abrogating its function. Lasty, Ochoa et al. [57] developed an antisenseoligonucleotide (ASO) which reversed pseudo-exon inclusion and restored the normal AIRE transcript sequence in LCLs. Ochoa et al. [57], concluded that:
• Their findings had revealed c.1504-818 G>A as a founder APECED-causing AIRE variant in the Puerto Rican population and uncovered pseudo-exon inclusion as an ASO-reversible genetic mechanism underlying APECED.
Melissa L Norton made an editorial summation related to the article in the same article [57] as follows:
• Biallelic mutations in the autoimmune regulator (AIRE) gene cause the autoimmune syndrome APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy).
• Here, Ochoa and colleagues had studied 17 patients, predominantly of Puerto Rican ancestry, who were clinically diagnosed as having APECED but who had lacked biallelic variants in AIRE exons or flanking intronic regions. They had identified a deep intronic variant in AIRE that created a cryptic splice site, leading to inclusion of a frame-shifting pseudo-exon which resulted in predicted alterations in the C terminus of the protein and loss of protein function.
• The authors developed an antisense oligonucleotide which restored normal AIRE splicing, suggesting a potential treatment approach for these patients.
• The Urofacial or Ochoa Syndrome (UFS or UFOS) is typified by an inverted facial expression whereby those afflicted by UFS seem to be crying while smiling which is associated with lower urinary tract dysfunction without evident obstructive or neurological cause.
• UFS is associated with autosomal recessive inheritance mutations in the HPSE2 gene, located at 10q23-q24, and the LRGI2 gene, located in 1p13.2; nevertheless, in up to 16% of patients, no associated mutations had been found.
• Recent evidence had indicated that these genes are critical to an adequate neurological development to the lower urinary tract and that the origin of the disease does appear to be related to the development of peripheral neuropathy.
• There is clinical variability among patients with UFS and not all patients afflicted by UFS manifest with the classic two components, and it had even been genetically confirmed in patients who had a prior diagnosis of Hinman Syndrome or other urinary bladder dysfunctions.
• Furthermore, manifestation of nocturnal lagophthalmos in these patients was recently reported.
• Clinical manifestation of UFS or UFOS is typified by pre-natal or childhood onset of urinary bladder voiding dysfunction, abnormal facial movement with [removed]resulting from abnormal co-contraction of the corners of the mouth and eyes), and often bowel dysfunction which entails constipation and/or encopresis.
• Urinary bladder voiding dysfunction could manifest prior to the birth of a baby in the form of mega-cystic urinary bladder.
• Within infancy and subsequent childhood, UFS could manifest with a poor urinary stream and dribbling urinary incontinence; incomplete emptying of the urinary bladder which could be ensued by the subsequent development of recurrent urinary tract infections, vesical-ureteric reflux, hydronephrosis and chronic kidney disease
• Clinical diagnosis of UFS Could be made in an individual who manifests with urinary tract dysfunction and typifying facial movement with expression, or the molecular diagnosis can be ascertained in an individual who manifests with the typifying features and biallelic pathogenic variants in either HPSE2 or LRIG2 identified based upon molecular genetic testing.
• It is important for clinicians to provide rapid and complete antibiotic medicament for cases of acute urinary tract infections.
• Anticholinergic and alpha-1 adrenergic blocking medicaments could respectively reduce urinary bladder (vesical) elevated pressure as well improve voiding of urine.
• Furthermore, the aforementioned medicaments could be aided by the undertaking of complemented by intermittent urethral catheterisation or by means of establishing a vesicostomy / suprapubic catheter insertion to drain the urine suprapubically
• When there is chronic kidney disease, the patients need to be referred to a nephrologist so that in the scenario of severe chronic cystic disease so that impaired kidney function options including dialysis which could be long-term peritoneal dialysis or haemodialysis or at times kidney transplantation could be considered if necessary
• Other treatments entail utilization of lubricant eye drops during the day and eye ointment nocte under the joint care of an ophthalmologist for nocturnal lagophthalmos as well as standard management for constipation and encopresis under the umbrella of a paediatric or adult gastroenterologist depending upon the age of the patient.
• Individual patients who are afflicted by UFS that are not yet complicated need to be looked after carefully by the multi-disciplinary management team with careful and regular surveillance assessments which should entail the ensuing:
Urinalysis to ascertain of the individual has developed urinary tract infection or not.
The undertaking of estimated glomerular filtration rate and renal function as base line assessments as well as follow-up assessments
The undertaking of ultrasound scan assessments of the urinary tract in order to assess the patients for complete or incomplete emptying of the urinary bladder as well as for hydronephrosis
Assessments of the patients for ocular involvement
Assessments by gastroenterologists for bowel dysfunction and usually at annual intervals unless there is a problem to be solved.
Patients need to be advised regarding the avoidance of nephrotoxic substances to avid development or worsening of chronic kidney disease.
It is important for clinicians to assess siblings of UFS afflicted individuals pursuant to their birth as soon as possible in order to ascertain whether or not they have facial and/or urinary tract presentations of UFS which would alert the clinician whether or not to assess the child for voiding dysfunction or renal function so as to commence any treatment that may be necessitated
There are no global consensus guidelines pertaining to a foetus who is considered to be at risk for the development of UFS to be managed pre-term,; nevertheless; the undertaking of ultrasound scan during the second as well as third trimester of the pregnant mother in order to ascertain if the baby in utero, has megacytis or hydronephrosis which would enable the multi-disciplinary team discuss as well agree on the timing of the delivery of the ne-born baby and if early delivery is required the delivery would then by undertaken in a tertiary referral centre with various specialists as well facilities for care of pre-term borne babies. Genetic counselling: UFS is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a UFS-related pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the UFS-related pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal and preimplantation genetic testing are possible.
• Early recognition and timely diagnosis of UFS are critical for prevention of complications emanating from UFS including recurrent urinary tract infections, hydronephrosis and chronic kidney disease
• Because of the rarity of UFS it is important for all clinicians globally to have a high index of the disease and to learn about the manifestations of UFS
• Prague Medical Report for granting copyright permission under the authors Skálová et al. [27] which permits reproduction of figures and contents of their journal article copyright permission which stated that “The journal applies the Creative Commons Attribution 4.0 International License to articles and other works we publish. If you submit your paper for publication by Prague Medical Report, you agree to have the CC BY license applied to your work. The journal allows the author(s) to hold the copyright without restrictions”.
• Archives of Clinical Nephrology for granting copyright permission for reproduction of figures and contents of their Journal article under copyright: © 2023 Barros Jacobino MN, et al. [38] This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
• International Journal of Rare Disease Disorders for granting Copyright permission for reproduction of figures and contents of their journal article under copyright: © 2021 Hazzab N, et al. [50] This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
None
Clearly Auctoresonline and particularly Psychology and Mental Health Care Journal is dedicated to improving health care services for individuals and populations. The editorial boards' ability to efficiently recognize and share the global importance of health literacy with a variety of stakeholders. Auctoresonline publishing platform can be used to facilitate of optimal client-based services and should be added to health care professionals' repertoire of evidence-based health care resources.

Journal of Clinical Cardiology and Cardiovascular Intervention The submission and review process was adequate. However I think that the publication total value should have been enlightened in early fases. Thank you for all.

Journal of Women Health Care and Issues By the present mail, I want to say thank to you and tour colleagues for facilitating my published article. Specially thank you for the peer review process, support from the editorial office. I appreciate positively the quality of your journal.

Journal of Clinical Research and Reports I would be very delighted to submit my testimonial regarding the reviewer board and the editorial office. The reviewer board were accurate and helpful regarding any modifications for my manuscript. And the editorial office were very helpful and supportive in contacting and monitoring with any update and offering help. It was my pleasure to contribute with your promising Journal and I am looking forward for more collaboration.

We would like to thank the Journal of Thoracic Disease and Cardiothoracic Surgery because of the services they provided us for our articles. The peer-review process was done in a very excellent time manner, and the opinions of the reviewers helped us to improve our manuscript further. The editorial office had an outstanding correspondence with us and guided us in many ways. During a hard time of the pandemic that is affecting every one of us tremendously, the editorial office helped us make everything easier for publishing scientific work. Hope for a more scientific relationship with your Journal.

The peer-review process which consisted high quality queries on the paper. I did answer six reviewers’ questions and comments before the paper was accepted. The support from the editorial office is excellent.

Journal of Neuroscience and Neurological Surgery. I had the experience of publishing a research article recently. The whole process was simple from submission to publication. The reviewers made specific and valuable recommendations and corrections that improved the quality of my publication. I strongly recommend this Journal.

Dr. Katarzyna Byczkowska My testimonial covering: "The peer review process is quick and effective. The support from the editorial office is very professional and friendly. Quality of the Clinical Cardiology and Cardiovascular Interventions is scientific and publishes ground-breaking research on cardiology that is useful for other professionals in the field.

Thank you most sincerely, with regard to the support you have given in relation to the reviewing process and the processing of my article entitled "Large Cell Neuroendocrine Carcinoma of The Prostate Gland: A Review and Update" for publication in your esteemed Journal, Journal of Cancer Research and Cellular Therapeutics". The editorial team has been very supportive.

Testimony of Journal of Clinical Otorhinolaryngology: work with your Reviews has been a educational and constructive experience. The editorial office were very helpful and supportive. It was a pleasure to contribute to your Journal.

Dr. Bernard Terkimbi Utoo, I am happy to publish my scientific work in Journal of Women Health Care and Issues (JWHCI). The manuscript submission was seamless and peer review process was top notch. I was amazed that 4 reviewers worked on the manuscript which made it a highly technical, standard and excellent quality paper. I appreciate the format and consideration for the APC as well as the speed of publication. It is my pleasure to continue with this scientific relationship with the esteem JWHCI.

This is an acknowledgment for peer reviewers, editorial board of Journal of Clinical Research and Reports. They show a lot of consideration for us as publishers for our research article “Evaluation of the different factors associated with side effects of COVID-19 vaccination on medical students, Mutah university, Al-Karak, Jordan”, in a very professional and easy way. This journal is one of outstanding medical journal.

Dear Hao Jiang, to Journal of Nutrition and Food Processing We greatly appreciate the efficient, professional and rapid processing of our paper by your team. If there is anything else we should do, please do not hesitate to let us know. On behalf of my co-authors, we would like to express our great appreciation to editor and reviewers.

As an author who has recently published in the journal "Brain and Neurological Disorders". I am delighted to provide a testimonial on the peer review process, editorial office support, and the overall quality of the journal. The peer review process at Brain and Neurological Disorders is rigorous and meticulous, ensuring that only high-quality, evidence-based research is published. The reviewers are experts in their fields, and their comments and suggestions were constructive and helped improve the quality of my manuscript. The review process was timely and efficient, with clear communication from the editorial office at each stage. The support from the editorial office was exceptional throughout the entire process. The editorial staff was responsive, professional, and always willing to help. They provided valuable guidance on formatting, structure, and ethical considerations, making the submission process seamless. Moreover, they kept me informed about the status of my manuscript and provided timely updates, which made the process less stressful. The journal Brain and Neurological Disorders is of the highest quality, with a strong focus on publishing cutting-edge research in the field of neurology. The articles published in this journal are well-researched, rigorously peer-reviewed, and written by experts in the field. The journal maintains high standards, ensuring that readers are provided with the most up-to-date and reliable information on brain and neurological disorders. In conclusion, I had a wonderful experience publishing in Brain and Neurological Disorders. The peer review process was thorough, the editorial office provided exceptional support, and the journal's quality is second to none. I would highly recommend this journal to any researcher working in the field of neurology and brain disorders.
Dear Agrippa Hilda, Journal of Neuroscience and Neurological Surgery, Editorial Coordinator, I trust this message finds you well. I want to extend my appreciation for considering my article for publication in your esteemed journal. I am pleased to provide a testimonial regarding the peer review process and the support received from your editorial office. The peer review process for my paper was carried out in a highly professional and thorough manner. The feedback and comments provided by the authors were constructive and very useful in improving the quality of the manuscript. This rigorous assessment process undoubtedly contributes to the high standards maintained by your journal.

International Journal of Clinical Case Reports and Reviews. I strongly recommend to consider submitting your work to this high-quality journal. The support and availability of the Editorial staff is outstanding and the review process was both efficient and rigorous.

Thank you very much for publishing my Research Article titled “Comparing Treatment Outcome Of Allergic Rhinitis Patients After Using Fluticasone Nasal Spray And Nasal Douching" in the Journal of Clinical Otorhinolaryngology. As Medical Professionals we are immensely benefited from study of various informative Articles and Papers published in this high quality Journal. I look forward to enriching my knowledge by regular study of the Journal and contribute my future work in the field of ENT through the Journal for use by the medical fraternity. The support from the Editorial office was excellent and very prompt. I also welcome the comments received from the readers of my Research Article.

Dear Erica Kelsey, Editorial Coordinator of Cancer Research and Cellular Therapeutics Our team is very satisfied with the processing of our paper by your journal. That was fast, efficient, rigorous, but without unnecessary complications. We appreciated the very short time between the submission of the paper and its publication on line on your site.

I am very glad to say that the peer review process is very successful and fast and support from the Editorial Office. Therefore, I would like to continue our scientific relationship for a long time. And I especially thank you for your kindly attention towards my article. Have a good day!

"We recently published an article entitled “Influence of beta-Cyclodextrins upon the Degradation of Carbofuran Derivatives under Alkaline Conditions" in the Journal of “Pesticides and Biofertilizers” to show that the cyclodextrins protect the carbamates increasing their half-life time in the presence of basic conditions This will be very helpful to understand carbofuran behaviour in the analytical, agro-environmental and food areas. We greatly appreciated the interaction with the editor and the editorial team; we were particularly well accompanied during the course of the revision process, since all various steps towards publication were short and without delay".

I would like to express my gratitude towards you process of article review and submission. I found this to be very fair and expedient. Your follow up has been excellent. I have many publications in national and international journal and your process has been one of the best so far. Keep up the great work.

We are grateful for this opportunity to provide a glowing recommendation to the Journal of Psychiatry and Psychotherapy. We found that the editorial team were very supportive, helpful, kept us abreast of timelines and over all very professional in nature. The peer review process was rigorous, efficient and constructive that really enhanced our article submission. The experience with this journal remains one of our best ever and we look forward to providing future submissions in the near future.

I am very pleased to serve as EBM of the journal, I hope many years of my experience in stem cells can help the journal from one way or another. As we know, stem cells hold great potential for regenerative medicine, which are mostly used to promote the repair response of diseased, dysfunctional or injured tissue using stem cells or their derivatives. I think Stem Cell Research and Therapeutics International is a great platform to publish and share the understanding towards the biology and translational or clinical application of stem cells.

I would like to give my testimony in the support I have got by the peer review process and to support the editorial office where they were of asset to support young author like me to be encouraged to publish their work in your respected journal and globalize and share knowledge across the globe. I really give my great gratitude to your journal and the peer review including the editorial office.

I am delighted to publish our manuscript entitled "A Perspective on Cocaine Induced Stroke - Its Mechanisms and Management" in the Journal of Neuroscience and Neurological Surgery. The peer review process, support from the editorial office, and quality of the journal are excellent. The manuscripts published are of high quality and of excellent scientific value. I recommend this journal very much to colleagues.

Dr.Tania Muñoz, My experience as researcher and author of a review article in The Journal Clinical Cardiology and Interventions has been very enriching and stimulating. The editorial team is excellent, performs its work with absolute responsibility and delivery. They are proactive, dynamic and receptive to all proposals. Supporting at all times the vast universe of authors who choose them as an option for publication. The team of review specialists, members of the editorial board, are brilliant professionals, with remarkable performance in medical research and scientific methodology. Together they form a frontline team that consolidates the JCCI as a magnificent option for the publication and review of high-level medical articles and broad collective interest. I am honored to be able to share my review article and open to receive all your comments.

“The peer review process of JPMHC is quick and effective. Authors are benefited by good and professional reviewers with huge experience in the field of psychology and mental health. The support from the editorial office is very professional. People to contact to are friendly and happy to help and assist any query authors might have. Quality of the Journal is scientific and publishes ground-breaking research on mental health that is useful for other professionals in the field”.

Dear editorial department: On behalf of our team, I hereby certify the reliability and superiority of the International Journal of Clinical Case Reports and Reviews in the peer review process, editorial support, and journal quality. Firstly, the peer review process of the International Journal of Clinical Case Reports and Reviews is rigorous, fair, transparent, fast, and of high quality. The editorial department invites experts from relevant fields as anonymous reviewers to review all submitted manuscripts. These experts have rich academic backgrounds and experience, and can accurately evaluate the academic quality, originality, and suitability of manuscripts. The editorial department is committed to ensuring the rigor of the peer review process, while also making every effort to ensure a fast review cycle to meet the needs of authors and the academic community. Secondly, the editorial team of the International Journal of Clinical Case Reports and Reviews is composed of a group of senior scholars and professionals with rich experience and professional knowledge in related fields. The editorial department is committed to assisting authors in improving their manuscripts, ensuring their academic accuracy, clarity, and completeness. Editors actively collaborate with authors, providing useful suggestions and feedback to promote the improvement and development of the manuscript. We believe that the support of the editorial department is one of the key factors in ensuring the quality of the journal. Finally, the International Journal of Clinical Case Reports and Reviews is renowned for its high- quality articles and strict academic standards. The editorial department is committed to publishing innovative and academically valuable research results to promote the development and progress of related fields. The International Journal of Clinical Case Reports and Reviews is reasonably priced and ensures excellent service and quality ratio, allowing authors to obtain high-level academic publishing opportunities in an affordable manner. I hereby solemnly declare that the International Journal of Clinical Case Reports and Reviews has a high level of credibility and superiority in terms of peer review process, editorial support, reasonable fees, and journal quality. Sincerely, Rui Tao.

Clinical Cardiology and Cardiovascular Interventions I testity the covering of the peer review process, support from the editorial office, and quality of the journal.

Clinical Cardiology and Cardiovascular Interventions, we deeply appreciate the interest shown in our work and its publication. It has been a true pleasure to collaborate with you. The peer review process, as well as the support provided by the editorial office, have been exceptional, and the quality of the journal is very high, which was a determining factor in our decision to publish with you.

The peer reviewers process is quick and effective, the supports from editorial office is excellent, the quality of journal is high. I would like to collabroate with Internatioanl journal of Clinical Case Reports and Reviews journal clinically in the future time.

Clinical Cardiology and Cardiovascular Interventions, I would like to express my sincerest gratitude for the trust placed in our team for the publication in your journal. It has been a true pleasure to collaborate with you on this project. I am pleased to inform you that both the peer review process and the attention from the editorial coordination have been excellent. Your team has worked with dedication and professionalism to ensure that your publication meets the highest standards of quality. We are confident that this collaboration will result in mutual success, and we are eager to see the fruits of this shared effort.

Dear Dr. Jessica Magne, Editorial Coordinator 0f Clinical Cardiology and Cardiovascular Interventions, I hope this message finds you well. I want to express my utmost gratitude for your excellent work and for the dedication and speed in the publication process of my article titled "Navigating Innovation: Qualitative Insights on Using Technology for Health Education in Acute Coronary Syndrome Patients." I am very satisfied with the peer review process, the support from the editorial office, and the quality of the journal. I hope we can maintain our scientific relationship in the long term.

Dear Monica Gissare, - Editorial Coordinator of Nutrition and Food Processing. ¨My testimony with you is truly professional, with a positive response regarding the follow-up of the article and its review, you took into account my qualities and the importance of the topic¨.

Dear Dr. Jessica Magne, Editorial Coordinator 0f Clinical Cardiology and Cardiovascular Interventions, The review process for the article “The Handling of Anti-aggregants and Anticoagulants in the Oncologic Heart Patient Submitted to Surgery” was extremely rigorous and detailed. From the initial submission to the final acceptance, the editorial team at the “Journal of Clinical Cardiology and Cardiovascular Interventions” demonstrated a high level of professionalism and dedication. The reviewers provided constructive and detailed feedback, which was essential for improving the quality of our work. Communication was always clear and efficient, ensuring that all our questions were promptly addressed. The quality of the “Journal of Clinical Cardiology and Cardiovascular Interventions” is undeniable. It is a peer-reviewed, open-access publication dedicated exclusively to disseminating high-quality research in the field of clinical cardiology and cardiovascular interventions. The journal's impact factor is currently under evaluation, and it is indexed in reputable databases, which further reinforces its credibility and relevance in the scientific field. I highly recommend this journal to researchers looking for a reputable platform to publish their studies.
Dear Editorial Coordinator of the Journal of Nutrition and Food Processing! "I would like to thank the Journal of Nutrition and Food Processing for including and publishing my article. The peer review process was very quick, movement and precise. The Editorial Board has done an extremely conscientious job with much help, valuable comments and advices. I find the journal very valuable from a professional point of view, thank you very much for allowing me to be part of it and I would like to participate in the future!”

Dealing with The Journal of Neurology and Neurological Surgery was very smooth and comprehensive. The office staff took time to address my needs and the response from editors and the office was prompt and fair. I certainly hope to publish with this journal again.Their professionalism is apparent and more than satisfactory. Susan Weiner

My Testimonial Covering as fellowing: Lin-Show Chin. The peer reviewers process is quick and effective, the supports from editorial office is excellent, the quality of journal is high. I would like to collabroate with Internatioanl journal of Clinical Case Reports and Reviews.

My experience publishing in Psychology and Mental Health Care was exceptional. The peer review process was rigorous and constructive, with reviewers providing valuable insights that helped enhance the quality of our work. The editorial team was highly supportive and responsive, making the submission process smooth and efficient. The journal's commitment to high standards and academic rigor makes it a respected platform for quality research. I am grateful for the opportunity to publish in such a reputable journal.

My experience publishing in International Journal of Clinical Case Reports and Reviews was exceptional. I Come forth to Provide a Testimonial Covering the Peer Review Process and the editorial office for the Professional and Impartial Evaluation of the Manuscript.

I would like to offer my testimony in the support. I have received through the peer review process and support the editorial office where they are to support young authors like me, encourage them to publish their work in your esteemed journals, and globalize and share knowledge globally. I really appreciate your journal, peer review, and editorial office.

Dear Agrippa Hilda- Editorial Coordinator of Journal of Neuroscience and Neurological Surgery, "The peer review process was very quick and of high quality, which can also be seen in the articles in the journal. The collaboration with the editorial office was very good."

I would like to express my sincere gratitude for the support and efficiency provided by the editorial office throughout the publication process of my article, “Delayed Vulvar Metastases from Rectal Carcinoma: A Case Report.” I greatly appreciate the assistance and guidance I received from your team, which made the entire process smooth and efficient. The peer review process was thorough and constructive, contributing to the overall quality of the final article. I am very grateful for the high level of professionalism and commitment shown by the editorial staff, and I look forward to maintaining a long-term collaboration with the International Journal of Clinical Case Reports and Reviews.

To Dear Erin Aust, I would like to express my heartfelt appreciation for the opportunity to have my work published in this esteemed journal. The entire publication process was smooth and well-organized, and I am extremely satisfied with the final result. The Editorial Team demonstrated the utmost professionalism, providing prompt and insightful feedback throughout the review process. Their clear communication and constructive suggestions were invaluable in enhancing my manuscript, and their meticulous attention to detail and dedication to quality are truly commendable. Additionally, the support from the Editorial Office was exceptional. From the initial submission to the final publication, I was guided through every step of the process with great care and professionalism. The team's responsiveness and assistance made the entire experience both easy and stress-free. I am also deeply impressed by the quality and reputation of the journal. It is an honor to have my research featured in such a respected publication, and I am confident that it will make a meaningful contribution to the field.

"I am grateful for the opportunity of contributing to [International Journal of Clinical Case Reports and Reviews] and for the rigorous review process that enhances the quality of research published in your esteemed journal. I sincerely appreciate the time and effort of your team who have dedicatedly helped me in improvising changes and modifying my manuscript. The insightful comments and constructive feedback provided have been invaluable in refining and strengthening my work".

I thank the ‘Journal of Clinical Research and Reports’ for accepting this article for publication. This is a rigorously peer reviewed journal which is on all major global scientific data bases. I note the review process was prompt, thorough and professionally critical. It gave us an insight into a number of important scientific/statistical issues. The review prompted us to review the relevant literature again and look at the limitations of the study. The peer reviewers were open, clear in the instructions and the editorial team was very prompt in their communication. This journal certainly publishes quality research articles. I would recommend the journal for any future publications.

Dear Jessica Magne, with gratitude for the joint work. Fast process of receiving and processing the submitted scientific materials in “Clinical Cardiology and Cardiovascular Interventions”. High level of competence of the editors with clear and correct recommendations and ideas for enriching the article.

We found the peer review process quick and positive in its input. The support from the editorial officer has been very agile, always with the intention of improving the article and taking into account our subsequent corrections.

My article, titled 'No Way Out of the Smartphone Epidemic Without Considering the Insights of Brain Research,' has been republished in the International Journal of Clinical Case Reports and Reviews. The review process was seamless and professional, with the editors being both friendly and supportive. I am deeply grateful for their efforts.

To Dear Erin Aust – Editorial Coordinator of Journal of General Medicine and Clinical Practice! I declare that I am absolutely satisfied with your work carried out with great competence in following the manuscript during the various stages from its receipt, during the revision process to the final acceptance for publication. Thank Prof. Elvira Farina

Dear Jessica, and the super professional team of the ‘Clinical Cardiology and Cardiovascular Interventions’ I am sincerely grateful to the coordinated work of the journal team for the no problem with the submission of my manuscript: “Cardiometabolic Disorders in A Pregnant Woman with Severe Preeclampsia on the Background of Morbid Obesity (Case Report).” The review process by 5 experts was fast, and the comments were professional, which made it more specific and academic, and the process of publication and presentation of the article was excellent. I recommend that my colleagues publish articles in this journal, and I am interested in further scientific cooperation. Sincerely and best wishes, Dr. Oleg Golyanovskiy.

Dear Ashley Rosa, Editorial Coordinator of the journal - Psychology and Mental Health Care. " The process of obtaining publication of my article in the Psychology and Mental Health Journal was positive in all areas. The peer review process resulted in a number of valuable comments, the editorial process was collaborative and timely, and the quality of this journal has been quickly noticed, resulting in alternative journals contacting me to publish with them." Warm regards, Susan Anne Smith, PhD. Australian Breastfeeding Association.

Dear Jessica Magne, Editorial Coordinator, Clinical Cardiology and Cardiovascular Interventions, Auctores Publishing LLC. I appreciate the journal (JCCI) editorial office support, the entire team leads were always ready to help, not only on technical front but also on thorough process. Also, I should thank dear reviewers’ attention to detail and creative approach to teach me and bring new insights by their comments. Surely, more discussions and introduction of other hemodynamic devices would provide better prevention and management of shock states. Your efforts and dedication in presenting educational materials in this journal are commendable. Best wishes from, Farahnaz Fallahian.

Dear Maria Emerson, Editorial Coordinator, International Journal of Clinical Case Reports and Reviews, Auctores Publishing LLC. I am delighted to have published our manuscript, "Acute Colonic Pseudo-Obstruction (ACPO): A rare but serious complication following caesarean section." I want to thank the editorial team, especially Maria Emerson, for their prompt review of the manuscript, quick responses to queries, and overall support. Yours sincerely Dr. Victor Olagundoye.

Dear Ashley Rosa, Editorial Coordinator, International Journal of Clinical Case Reports and Reviews. Many thanks for publishing this manuscript after I lost confidence the editors were most helpful, more than other journals Best wishes from, Susan Anne Smith, PhD. Australian Breastfeeding Association.

Dear Agrippa Hilda, Editorial Coordinator, Journal of Neuroscience and Neurological Surgery. The entire process including article submission, review, revision, and publication was extremely easy. The journal editor was prompt and helpful, and the reviewers contributed to the quality of the paper. Thank you so much! Eric Nussbaum, MD
Dr Hala Al Shaikh This is to acknowledge that the peer review process for the article ’ A Novel Gnrh1 Gene Mutation in Four Omani Male Siblings, Presentation and Management ’ sent to the International Journal of Clinical Case Reports and Reviews was quick and smooth. The editorial office was prompt with easy communication.

Dear Erin Aust, Editorial Coordinator, Journal of General Medicine and Clinical Practice. We are pleased to share our experience with the “Journal of General Medicine and Clinical Practice”, following the successful publication of our article. The peer review process was thorough and constructive, helping to improve the clarity and quality of the manuscript. We are especially thankful to Ms. Erin Aust, the Editorial Coordinator, for her prompt communication and continuous support throughout the process. Her professionalism ensured a smooth and efficient publication experience. The journal upholds high editorial standards, and we highly recommend it to fellow researchers seeking a credible platform for their work. Best wishes By, Dr. Rakhi Mishra.

Dear Jessica Magne, Editorial Coordinator, Clinical Cardiology and Cardiovascular Interventions, Auctores Publishing LLC. The peer review process of the journal of Clinical Cardiology and Cardiovascular Interventions was excellent and fast, as was the support of the editorial office and the quality of the journal. Kind regards Walter F. Riesen Prof. Dr. Dr. h.c. Walter F. Riesen.

Dear Ashley Rosa, Editorial Coordinator, International Journal of Clinical Case Reports and Reviews, Auctores Publishing LLC. Thank you for publishing our article, Exploring Clozapine's Efficacy in Managing Aggression: A Multiple Single-Case Study in Forensic Psychiatry in the international journal of clinical case reports and reviews. We found the peer review process very professional and efficient. The comments were constructive, and the whole process was efficient. On behalf of the co-authors, I would like to thank you for publishing this article. With regards, Dr. Jelle R. Lettinga.

Dear Clarissa Eric, Editorial Coordinator, Journal of Clinical Case Reports and Studies, I would like to express my deep admiration for the exceptional professionalism demonstrated by your journal. I am thoroughly impressed by the speed of the editorial process, the substantive and insightful reviews, and the meticulous preparation of the manuscript for publication. Additionally, I greatly appreciate the courteous and immediate responses from your editorial office to all my inquiries. Best Regards, Dariusz Ziora

Dear Chrystine Mejia, Editorial Coordinator, Journal of Neurodegeneration and Neurorehabilitation, Auctores Publishing LLC, We would like to thank the editorial team for the smooth and high-quality communication leading up to the publication of our article in the Journal of Neurodegeneration and Neurorehabilitation. The reviewers have extensive knowledge in the field, and their relevant questions helped to add value to our publication. Kind regards, Dr. Ravi Shrivastava.

Dear Clarissa Eric, Editorial Coordinator, Journal of Clinical Case Reports and Studies, Auctores Publishing LLC, USA Office: +1-(302)-520-2644. I would like to express my sincere appreciation for the efficient and professional handling of my case report by the ‘Journal of Clinical Case Reports and Studies’. The peer review process was not only fast but also highly constructive—the reviewers’ comments were clear, relevant, and greatly helped me improve the quality and clarity of my manuscript. I also received excellent support from the editorial office throughout the process. Communication was smooth and timely, and I felt well guided at every stage, from submission to publication. The overall quality and rigor of the journal are truly commendable. I am pleased to have published my work with Journal of Clinical Case Reports and Studies, and I look forward to future opportunities for collaboration. Sincerely, Aline Tollet, UCLouvain.

Dear Ms. Mayra Duenas, Editorial Coordinator, International Journal of Clinical Case Reports and Reviews. “The International Journal of Clinical Case Reports and Reviews represented the “ideal house” to share with the research community a first experience with the use of the Simeox device for speech rehabilitation. High scientific reputation and attractive website communication were first determinants for the selection of this Journal, and the following submission process exceeded expectations: fast but highly professional peer review, great support by the editorial office, elegant graphic layout. Exactly what a dynamic research team - also composed by allied professionals - needs!" From, Chiara Beccaluva, PT - Italy.

Dear Maria Emerson, Editorial Coordinator, we have deeply appreciated the professionalism demonstrated by the International Journal of Clinical Case Reports and Reviews. The reviewers have extensive knowledge of our field and have been very efficient and fast in supporting the process. I am really looking forward to further collaboration. Thanks. Best regards, Dr. Claudio Ligresti
Dear Chrystine Mejia, Editorial Coordinator, Journal of Neurodegeneration and Neurorehabilitation. “The peer review process was efficient and constructive, and the editorial office provided excellent communication and support throughout. The journal ensures scientific rigor and high editorial standards, while also offering a smooth and timely publication process. We sincerely appreciate the work of the editorial team in facilitating the dissemination of innovative approaches such as the Bonori Method.” Best regards, Dr. Matteo Bonori.

I recommend without hesitation submitting relevant papers on medical decision making to the International Journal of Clinical Case Reports and Reviews. I am very grateful to the editorial staff. Maria Emerson was a pleasure to communicate with. The time from submission to publication was an extremely short 3 weeks. The editorial staff submitted the paper to three reviewers. Two of the reviewers commented positively on the value of publishing the paper. The editorial staff quickly recognized the third reviewer’s comments as an unjust attempt to reject the paper. I revised the paper as recommended by the first two reviewers.
