
Review Article | DOI: https://doi.org/10.31579/JCRA/003Copy
*Corresponding Author: ingping Wang, Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital, 55 Fruit Street, Boston, USA. E
Citation: Melanie MS and Jingping W., (2019) Central Nervous System Toxicity during the Induction of General Anesthesia After Intravenous Bolus of Lidocaine. International Journal of Mediators of Inflammation, 2(1); DOI: 10.31579/JCRA/003
Copyright: ©2019. Jingping W. This is an open-access article distributed under the termsof the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 11 November 2019 | Accepted: 12 December 2019 | Published: 19 December 2019
Keywords: lidocaine; central nervous system (cns); cardiovascular toxicity
Lidocaine is a frequently utilized intravenous adjuvant for the induction of general anesthesia. Despite its wide therapeutic margin of safety, central nervous system (CNS) and cardiovascular toxicity may ensue. A healthy, 22-year-old female presented for a temporomandibular joint (TMJ) arthroscopy and manipulation. After a 2 mg kg-1 intravenous bolus of lidocaine, the patient developed symptoms of lidocaine CNS toxicity. This paper will overview the pharmacology, structure and mechanism of lidocaine, the recognition and management of systemic lidocaine toxicity, and the patient specific contributors to the development of lidocaine toxicity. Subsequently, innovative practice recommendations will be discussed.
Objectives
Lidocaine is a frequently utilized intravenous adjuvant for the induction of general anesthesia. 15 Despite its wide therapeutic margin of safety, central nervous system (CNS) and cardiovascular toxicity may ensue. 23 Vigilance is essential in the recognition and immediate treatment of suspected systemic lidocaine toxicity. We present a case of CNS toxicity after an intravenous bolus of lidocaine during the induction of general anesthesia.
Case Presentation
A 22-year-old female presented for a temporomandibular joint (TMJ) arthroscopy and manipulation. The patient’s pertinent medical history included orofacial pain and daily migraines refractory to other treatment modalities including thoracenteses, physical therapy and use of a night guard. The patient had a past medical history significant for Chiari 1 malformation, intractable migraines with aura, tethered spinal cord, seasonal allergies, mild asthma, gastroesophageal reflux, MVA, and post-operative nausea and vomiting.
The patient’s home medication list included: acetaminophen 325 mg tablet, albuterol 90 mcg/actuation inhaler, almotriptan 12.5 MG tablet, aspirin-acetaminophen-caffeine 250-250-65 mg per tablet, botulinum toxin type A 100 unit SolR, cetirizine 10 MG tablet, fluticasone propionate 50 mcg/actuation nasal spray, ibuprofen 200 MG tablet, levonorgestrel 14 mcg/24 hour, melatonin 10 mg tablet, oxycodone 5 MG tablet, and tramadol 50 mg tablet. Prior to the induction of anesthesia, the patient received 20 mg of intravenous famotidine, a transdermal 1.5 mg scopolamine patch, and an oral 975 mg dosage of acetaminophen.
During induction of anesthesia, the patient developed anxiety, tinnitus, a foul taste in the mouth, and tremors immediately after receiving a dose of lidocaine at a 2 mg kg-1. Patient was then given 2 mg morphine and 150 mg propofol followed by a prophylactic 2 mg intravenous bolus of midazolam. Subsequently, the patient received 80 mg of succinylcholine for intubation. A 6.0 mm Nasal Rae tube was successfully placed and confirmed via fiberoptic bronchoscopy. The patient was administered an FiO2 of 100%, inhaled volatile anesthetic (sevoflurane) and hyperventilated to an ETCO2 of 30 mmHg. The notable hemodynamic changes after intubation were tachycardia (HR=167 BPM) and hypertension (BP=150/85; MAP=106mmHg), which were corrected with a 10 mg bolus of IV esmolol. The patient’s ventilation was maintained with a target ETCO2 of 30-32 mmHg throughout the case.
The patient’s cardiovascular status remained stable throughout the duration of the procedure. The patient received no further administration of lidocaine by the surgical or anesthesia team. There were no post-operative complications.
Lidocaine: Pharmacology and Clinical Indications
Lidocaine is a frequently utilized intravenous adjuvant for the induction of general anesthesia. 15 Lidocaine’s molecular structure consists of a hydrophilic diethylglycine molecule bound via an amide bond to a hydrophobic aromatic benzene ring. 6 The amphipathic nature of lidocaine permits permeability across both hydrophobic and hydrophilic membranes. 22 Lidocaine metabolism is hepatic, via cytochrome P450 3A4. 5 Lidocaine elimination half-life is 1.5- 2 hour with renal excretion. 3, 6
The molecular structure of lidocaine (2-diethylaminoaceto-2’,6’-xylidide) 17 is displayed below:
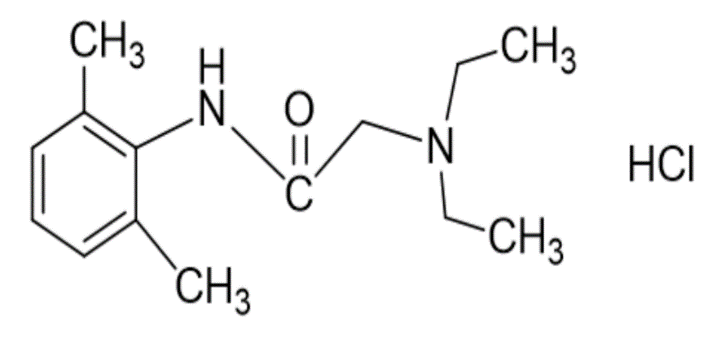
Sodium channel antagonists, such as lidocaine, inhibit ectopic discharges of TTX-sensitive and TTX-resistant sodium channels without affecting nerve conduction. 15 Sodium channel blockade results in a reduction of neuronal action potential initiation, propagation and facilitates membrane stabilization. 26 The peak plasma concentrations (Cmax) obtained from a 1.5-2 mg/kg bolus of intravenous lidocaine inhibit a small number of sodium channels, thus secondary and tertiary mechanisms are likely involved. In progressively higher serum concentrations, lidocaine will inhibit voltage-gated potassium channels, voltage-gated calcium channels and nicotinic acetylcholine (nAch) receptors. 22
Intravenous lidocaine has been shown to target polymorphonuclear granulocytes by interfering with upstream G-protein pro-inflammatory signaling and the release of reactive oxygen species and cytokines. 7 Intravenous lidocaine results in a reduction of cerebral metabolic rate, cerebral blood flow, and cerebral metabolic oxygen consumption, thus, reducing energy required for neuronal membrane stabilization. 1,15 The CNS inhibitory effects of systemic lidocaine may be influenced by lidocaine mediated strychnine-sensitive glycine receptors of wide dynamic range neurons in the spinal cord. 4 Lidocaine is also shown to block the K+-Cl− cotransporter (KCC) on γ-aminobutyric acid (GABAA) receptors. 16 This blockade and lidocaine mediated inhibition likely plays a role in the progressive nervous system symptoms and seizure activity observed in systemic lidocaine CNS toxicity. 29
N-methyl-D-aspartate (NMDA) receptors play an important role in nociception via fast excitatory neurotransmission in the CNS 24. Lidocaine plays a role in reducing hyperalgesia by its effects on NR1/2A NMDA receptor subunits.11 Lidocaine is shown to indirectly inhibit NMDA via its effects on protein kinase C (PKC) in a concentration dependent manner. 11 Lidocaine inhibits PKC mediated functions intracellularly, resulting in a reduction of PKC phosphorylation of NMDA subunits, consequently, reducing downstream signaling and NMDA receptor function. 11
A 1.5 mg kg-1 bolus of intravenous lidocaine during anesthetic induction serves a number of purposes: the attenuation of pain response to propofol and/or etomidate administration, blunting the cough reflex elicited by a rapid fentanyl bolus and airway instrumentation, reduction of succinylcholine-induced elevations in intragastric pressure and ICP, mitigating the hemodynamic response (hypertension and tachycardia) from laryngoscopy, blunting increases in ICP secondary to airway instrumentation, and to mitigate airway reactivity. 8, 13, 14, 15, 18, 27, 30 Systemic lidocaine has also been utilized for its analgesic, anti-inflammatory properties, and its synergistic reduction in the minimum alveolar concentration (MAC) of volatile anesthetics by 10-28%. 9, 27

The plasma concentration of lidocaine is dependent upon several factors including: dosage, absorption, distribution, elimination, patient age, patient hepatic function, patient cardiovascular status, and plasma protein binding. 3 Lidocaine injection can be illustrated by a two-compartment model resulting in an initial rapid reduction of peak plasma concentrations (Cmax). 3, 23 The primary α-phase includes rapid uptake to the lungs and distribution to the highly vascular compartment. 3, 23 The subsequent β-phase results in a slow re-distribution to vascular poor regions. 3, 23 The vasodilatory effects of lidocaine contribute to its short duration of action and enhanced systemic absorption. 23
Lidocaine’s high hepatic extraction ratio (0.63) makes it dependent on hepatic blood flow. 21 The metabolism of lidocaine is dependent upon CYP450 34A enzyme function and hepatocellular perfusion. 3 The hepatic metabolism of lidocaine occurs via CYP450 34A enzyme N-dealkylation and hydroxylation. 3 The CYP450 34A metabolism results in the formation of monoethylglycinexylidide via hepatic oxidative dealkylation and subsequently the formation of glycine xylidine via hydroxylation. 3,23 The elimination half-life (t 1/2/β) of lidocaine is 1.5-2 hours and excretion of metabolites occurs via the kidney with <5% excreted unmetabolized3.
Factors that may influence the unbound free fraction of systemic lidocaine include: a reduction in plasma protein binding, increased total interstitial volume, and reduced total plasma volume. 21 The unbound free fraction of systemic lidocaine is responsible for its therapeutic effects. 5 Increases in unbound free fraction of lidocaine may be a result of physiological states such as: pregnancy, cirrhosis, and newborn age. 3
Central Nervous System Toxicity
Systemic lidocaine toxicity is a life-threatening emergency that begins with CNS symptoms, and if left untreated, may progress to cardiovascular toxicity and fatality. Lidocaine toxicity is dose dependent and largely influenced by Cmax. 3 Cmax is directly influenced by the free fraction of lidocaine available. 5 The free fraction of lidocaine is contingent upon plasma protein binding, total interstitial volume, and plasma volume. 21 The extent of plasma protein binding is influenced by the serum pH and patient levels of alpha 1-glycoprotein and albumin. 3 There is an indirect relationship between seizure threshold and the levels of serotonin (5-hydroxytryptophan) in the central nervous system. 23 Thus, medications that elevated levels of 5-hydroxytryptophan may increase the risk for lidocaine CNS toxicity. 23
At a Cmax of 3-4 mcg mL-1 patients develop circumoral numbness, restlessness, tinnitus, tingling of the face, metallic taste in the mouth, auditory hallucinations, and visual hallucinations. 3, 20, 22 At a Cmax of 5-10 mcg mL-1 tremors and seizure activity may ensue. 3, 20, 22 Thus, intravenous lidocaine boluses should be titrated to a serum concentration less than 5-10 mcg mL-1, the seizure threshold for lidocaine. 15, 22 If toxicity remains untreated, it may progress to bradycardia, arrhythmia, hypotension, cardiac and respiratory arrest. 22
Management of Central Nervous System Toxicity
The initial management of lidocaine toxicity includes the following steps: call for back-up clinical support, stop lidocaine administration, intubation to secure the airway and administer a benzodiazepine. 22 In a patient with no cardiovascular symptoms, propofol can also be administered at a 1 mg kg-1 IV. 22 A 20% lipid emulsion may be administered at a 1.5 mL kg-1 followed by a continuous infusion of 0.25-0.5 mL kg-1 min-1. 22 If the toxicity progresses to cardiovascular compromise, ACLS should be followed with the following modifications: use 10-100 mcg doses of epinephrine, utilize amiodarone for ventricular arrhythmias, avoid vasopressin, calcium channel blockers, beta-blockers, and consider cardiopulmonary bypass for patients that do not respond to treatment. 22
The possible explanations for CNS toxicity in this case include: pharmaceutical interactions, cytochrome P450 3A4 mediated drug-drug interactions, concurrent beta-blockade, rate of administration and dosage.
Practice Recommendations
Notes
The authors declare that no competing interests exist.
Ethical Declarations
Informed consent was obtained from the patient in this case study. All associated patient identifiers were removed for the purposes of this paper.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
